INVESTIGATING THE CELLULAR MACHINERY: PROTEIN IDENTIFICATION
Proteins are the molecules that are responsible for the majority of functions inside the cell. Thus scientists are usually most interested in which proteins and how much of each protein are present in a cell following a particular experimental manipulation. Unfortunately, it is difficult to study global protein expression, since proteins cannot be replicated easily in the lab. Also, proteins do not have complementary sequences like DNA or RNA that can be used as probes. For this reason, many scientists rely on gene expression analysis to give clues as to how a protein is being regulated. However, new technologies that reliably determine which proteins are present in the cell and at what level is making the task of studying proteins much more accessible. The study of proteins is known as proteomics.
Studying proteins requires two stages of sample preparation. Proteins are separated using 2-dimensional electrophoresis, followed by identification using mass spectrometry (MS).
Protein Separation
2-dimensional electrophoresis separates proteins based on size, as in regular electrophoresis, but also based on charge, or isoelectric point (pI). The first step is isoelectric focusing (IEF). The mixed protein sample is run on an immobilized pH gradient, the range of the gradient used depends on the expected proteins in the sample. The sample is added to the gradient and an electric current is applied. Proteins will be positively charged at pH’s below their pI and negatively charged at pH’s above their pI. When the protein is at the point in the gradient where the surrounding pH is equal to it’s pI, there will be no charge on the protein and it will stop moving.
Once enough time has passed for the proteins to settle in the gradient, the current is removed and the gradient is laid horizontally along an SDS-PAGE gel. An electric current is then applied and the proteins move horizontally out of the IEF gradient and into the polyacrylamide gel where they are separated based on molecular weight. This method can reproducibly separate mixtures of proteins. An example of a 2-D gel is shown in Figure 1.
Once the proteins have been separated, they can be analyzed quantitatively as long as there is a reference sample. The amount of protein in cells under two conditions (e.g. aerobic and anaerobic) can be measured by staining with a fluorescent dye. The brighter the fluorescence, the more protein is present. Proteins that are expressed at different levels are then taken for further analysis and identification.

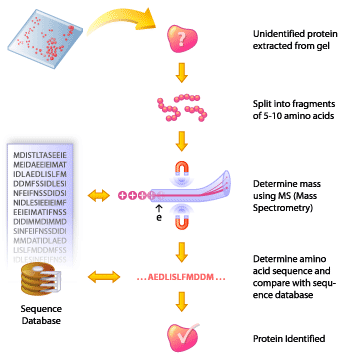
Protein Identification by Mass Spectrometry
The spots are cut out of the gel and digested into smaller polypeptide fragments (5 to 10 amino acids) by enzymes. The polypeptide fragments are analyzed by mass spectrometry, which will give the molecular weight of each fragment. Once the masses of the fragments have been determined, they are run through a sequence database and compared to find the actual amino acid sequence, and thus identity, of the protein.
This method of protein analysis also gives information on any post-translational modifications that have occurred, such as alternate splicing, glycosylation or phosphorylation.
Monitoring expression by this method is useful for measuring levels of active proteins in a cell and may give clues to the metabolism, signaling or other activities of a cell under varying conditions.
Further Applications
Another important component of proteome analysis is the interactions between proteins. Proteins can act as messengers, binding to other protein receptors to send a signal to the cell, protein-protein interactions can form structural connections between cells, and some enzyme inhibitors are proteins. While there are many ways to test interactions between two specific proteins (yeast two-hybrid system, tandem affinity purification, enzyme inhibition assays) it is still difficult to detect many interactions at once.
A protein chip, similar to the microarray, is under development that would be a huge breakthrough in proteome analysis. A chip that held thousands of immobilized proteins could be used to detect protein-protein interactions quickly and easily.
MacBeath and Schreiber published a paper in Science of 2000 describing the successful printing of proteins as a microarray [1]. The proteins had to be spotted on the array while retaining their original, active shape and also had to remain in solution to stay active. The researchers accomplished this and demonstrated three known interactions, including Protein G and Immunoglobulin G, could be detected by this method.
Biacore and Ciphergen are the two companies that currently produce protein chips however many companies are expected to enter the market once they have resolved the technical difficulties of ensuring that each one of the thousands of immobilized proteins must remain active and stable for a reasonable amount of time.
References
1. MacBeath G., Schreiber S. (2000). Printing proteins as microarrays for high-throughput function determination. Science 289(5485):1760-3.
(Art by Jiang Long – note that high res versions of image files available here)