CREATINE: FROM MUSCLE TO BRAIN
Creatine is an organic molecule which was discovered in the first half of the nineteenth century, in meat extract resembling beef broth. A century later, in the first half of the twentieth century, another substance was found, this time in frog extract, being phosphocreatine – or creatine with a phosphate group stuck on it. It turned out that creatine and phosphocreatine are part of an intracellular energy management system, and the two have had a remarkable history since. I’m not alluding to the fact that around 2.5 million kg of creatine are consumed yearly by athletes in the belief that it furthers their physical endurance and strength [1]. Instead, I’m alluding to the fact that creatine appears to be far more important for the brain than for muscle, and that individuals deficient in creatine, suffer from severe intelligence deficits, speech problems, and epilepsy. Underlying this story are insights from the physiology and biochemistry of creatine, which involve astute clinical observations, cutting edge technology, genetic analysis and epidemiological data, all ultimately suggesting that a particular form of creatine deficiency may be a major cause of mental retardation in boys. In this paper, I shall touch briefly on each of these points to illustrate recent developments in this exciting branch of metabolic medicine.
The Biochemical Background
Cells need to maintain a constant energy flux in order to stay alive. Energy is handled within cells in the form of high energy phosphate bonds. The principle behind this is that some chemical bonds need a lot of energy to be welded. Adding one phosphate group (P) to adenosine-mono-phosphate creates such a high energy bond. The resulting compound is adenosine-di-phosphate, or ADP. Repeat this, and you get another high energy bond, the compound being called by analogy adenosine-tri-phosphate or ATP. Conversely, if you have an ATP and remove one P, you get ADP, P, and some energy, which in turn may be used for all sorts of energy requiring tasks, like ion pumping or contracting special proteins. The energy for forming ADP or ATP stems from the intracellular degradation of foodstuffs like carbohydrates or fats, which for the most part takes place in mitochondria.
Overall, the picture is that of a cycle, where ADP and P are welded together with energy provided mainly by mitochondria. ATP is then brought to energy consuming machinery, an ion pump say, the energy is consumed, ATP is broken down into ADP and P, and both return to mitochondria in order to store another bucket full of energy.
So where does creatine come into the picture? Creatine (Cr) is also a substance that can form high energy phosphate bonds. The resulting compound is called phosphocreatine (PCr), and certain cell types also maintain intracellular Cr and PCr pools in addition to ADP and ATP. Muscle cells and neurons are two such cell types. Why might they do this? Neurons and muscle cells both have large and fluctuating energy requirements. Muscles need to be able to contract repeatedly on command, when running for a bus, for instance. Neurons need the potential to fire away suddenly and sustainably, as in when viewing a beautiful painting for instance – this requires a lot of ion pumping to maintain electrical potentials. In order to alleviate the strain of abrupt energy demands on the ATP-ADP system, these cells use a backup, which is the PCr-Cr system.
In essence, creatine can take over a high energy phosphate group from ATP and store it as PCr. Conversely, it may return a high energy phosphate to ADP, generating one ATP. This capacity allows the cell to store additional energy, increase ATP generation on a short term basis, and buffers the effects of a sudden high demand for ATP. Another advantage of having a PCr-Cr system is apparent from a comparison of the sizes of adenosine vs. creatine. Adenosine is large, creatine is small. Large molecules have a hard time traversing the distance between mitochondria and ion pumps within the cell, small molecules are much better for this. For this reason, PCr is more effective as transport vehicle for high energy bonds than ATP [2]. It is this involvement in the cellular energy metabolism that has led to the idea that getting more creatine into muscles will improve strength and endurance, which, incidentally is true mostly for exercises associated with repeated bouts of activity [1].
How does creatine get into muscle and brain? Creatine is not synthesised in either muscle cells or neurons, but is rather taken up from the fluid surrounding these cells. This uptake is effected by a special carrier, the creatine transporter, or CreaT for short. But how does creatine get into the surrounding fluid in the first place? There are two ways. First, creatine is contained in the food we eat, especially in meat. The meat is digested and creatine is selectively picked out by the gut and transferred to the bloodstream, which subsequently delivers it to muscles and the brain. However, creatine is also synthesised in the body, mainly in the kidney and liver. This is quantitatively more important and I shall thus dwell on it a little more.
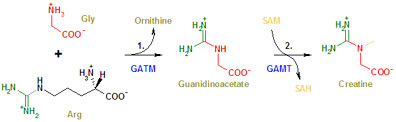
Creatine is synthesised by a two step process. In a first step, two amino acids (= constituents of proteins), arginine and glycine, are used to create an intermediary product called guanidinoacetic acid. This is also found in the bloodstream. In a second step, guanidinoacetic acid reacts with a methyl group donated by a molecule called SAM (never mind the details, for now), resulting in creatine. These steps are catalysed by two specific enzymes, the first is called GATM (Guanidinoacetate N-methyltransferase, also known as AGAT or L-arginine:glycine amidinotransferase), the second is called GAMT (or S-adenosyl-L-methionine:N-guanidinoacetate methyltransferase) (Figure 1) [3]. This will become important because all three mediators of creatine metabolism in the body, CreaT, AGAT, and GAMT may be impaired on the basis of defective genes coding for them. Creatine degrades spontaneously after a while to creatinine which leaves the cells, enters the bloodstream and is excreted by the kidneys.
In the Hospital
Insight into the importance of creatine in the brain began in 1994: here, a patient was presented to a paediatric clinic in Goettingen, Germany. In fact, he was specifically diagnosed with a deficiency of creatine in the brain [4]. The boy was well until 4 months of age, at which point his development stopped, his muscles lost tension, his head started nodding, and he started to have uncontrollable movements that were both jerky and almost violent. At 22 months he was unable to sit or roll over, and uncoordinated swallowing made eating difficult. Biochemical investigations revealed very low plasma levels of creatinine, the degradation product of creatine. Yet, the symptoms of this patient seemed largely due to some malfunction of the brain. None of the diagnoses known at the time fitted this picture.
An important tool to pin down the cause for this boy’s neurological symptoms was an investigation using a technique known as nuclear magnetic resonance spectroscopy (NMR spectroscopy). This procedure harnesses the fact that some types of atoms in a given molecule have rotating nuclei, for instance protons. Rotating and positively charged nuclei generate a magnetic field of their own – this is one of the principle tenets of electromagnetic theory. Imagine if you place a magnet in an external magnetic field, it will attempt to align itself with this field. If you further imagine that this magnet is suspended somehow in an external magnetic field, and that it has aligned itself successfully with the field, and then you bang it somehow, what happens? It will oscillate until it is aligned again. The frequency of oscillation is related to the specific characteristics of the magnet attempting alignment.
Therefore, the principle of nuclear magnetic spectroscopy is to place a sample into a magnetic field, bang the nuclei with an electromagnetic impulse, and measure their frequencies of oscillation by detecting electromagnetic waves emitted during the course of the oscillations. In the case of nuclei, this is complicated by the fact that they are spinning, but regardless, these frequencies are analysed as energy radiated at a given frequency, i.e. a spectrum. The oscillation frequencies of the nuclei are determined by the type of nucleus you are looking at and by the electromagnetic microenvironment it finds itself in, i.e. the chemical bonds and interactions with other atoms nearby (chemical shift of the frequencies). This allows you to detect signals from specific molecules in your sample. In our case, nuclear magnetic resonance spectroscopy which looks at hydrogen nuclei, i.e. protons, allows us to quantify creatine concentrations in the brain.
In this manner, proton NMR spectroscopy was used to investigate the brain of the above patient and revealed the complete absence of creatine. This raises three questions. Firstly, why is there no creatine? Secondly, can the lack of creatine explain the symptoms? And thirdly, can his condition be improved by providing him with additional creatine? A hint for the answer to the first question was found in the fact that the concentration of guanidinoacetate was elevated in the brain of the patient.
Looking at the pathway for creatine biosynthesis (Figure 1), this suggests that GAMT may not be working properly, and guanidinoacetate accumulates because it can no longer be processed. Genetic analysis indeed confirmed that the patient had two mutant GAMT alleles, i.e. no functional gene for this enzyme [5]. The whole situation suggested that creatine synthesis in the organism is important for brain function and development. However, the reason for this is still unknown. Hypotheses range from an important contribution of the PCr-Cr system to axonal growth to a general impairment of energy metabolism in the brain. So the answer to the second question is still obscure. In passing it needs to be mentioned that raised guanidinoacetate levels in brain can also cause some aspects of the overall menu of symptoms. As to the third question: Creatine supplementation led to a remarkable improvement of the symptoms. After two months on creatine the infant’s jerky movements had disappeared, he started to develop again, he learned to sit and to crawl, and at 4.5 years he was able to walk. Creatine levels in the brain increased in parallel with these improvements [6].
The Biochemical Defects and Their Scope
Since this first description of a patient with creatine deficiency in the brain (due to a defect of the GAMT enzyme), other patients have been diagnosed with creatine deficiency stemming from defects of AGAT [7], (which catalyses the first step of creatine synthesis), and defects of the creatine transporter CreaT [8]. Each of these pathologies results in depleted creatine stores in the brain. Clinical hallmarks are now recognised to be mental retardation, expressive speech delay, and epilepsy.
The biochemical abnormalities vary, however, due to differing mechanisms for creatine deficiency on a molecular level. For instance, in GAMT deficient patients guanidinoacetate accumulates in blood and brain. Guanidinoacetate is toxic to the brain and aggravates the symptoms caused by creatine deficiency alone. Patients with a lack of AGAT activity appear to have very low guanidinoacetate levels. Patients with a defective creatine transporter, on the other hand, are fully able to synthesise enough creatine for their needs, but they appear to be unable to transport it into the brain. The levels of creatine, creatinine, and guanidinoacetic acid in the blood of these patients is normal. For this instance, and given that the defect resides in the transport, additional supplementation of creatine seems to be doomed as a therapy in these cases – indeed, it is notoriously ineffective.
Genetically, both the AGAT and GAMT genes are autosomal and the respective diseases are inherited in an autosomal recessive pattern. In contrast, the gene for CreaT is on the X chromosome and thus it is inherited in an X-linked manner, whereby males are affected almost exclusively with their mothers being carriers. Consequently, CreaT defects stand out in another respect, too: Whereas the total number of patients known with defective GAMT is 27, and the total number for AGAT is 4, there are currently more than 50 patients known with CreaT defects.
Since the disease has only recently been identified, this cannot be taken as an estimate of the real frequency, since screening and diagnostic possibilities are only just being implemented in most centers. A recent study based on the frequencies of individual mutations of the CreaT gene has estimated that as much as 2% of all cases of X-linked mental retardation are caused by a defect in the creatine transporter [9]. Most patients with mental retardation remain undiagnosed, to date, and of these, a considerable proportion are male, with the overhang likely due to X-linked diseases causing mental retardation. So if the estimates by Rosenberg et al. are accurate, a treatment for CreaT deficiency could mean a substantive progress for many of these patients. Still, it is unfortunate that the most frequent of the creatine deficiency syndromes is at the same time the most untreatable at present. Some ingenuity will be required to find a way to supply creatine to neurons which lack an operative creatine transporter.
References
[1] R.L. Terjung, P. Clarkson, E.R. Eichner, P.L. Greenhaff, P.J. Hespel, R.G. Israel, W.J. Kraemer, R.A. Meyer, L.L. Spriet, M.A. Tarnopolsky, A.J. Wagenmakers, M.H. Williams, American College of Sports Medicine roundtable. The physiological and health effects of oral creatine supplementation, Med Sci Sports Exerc 32 (2000) 706-717.
[2] S.P. Bessman, C.L. Carpenter, The creatine-creatine phosphate energy shuttle, Annu Rev Biochem 54 (1985) 831-862.
[3] J.B. Walker, Creatine: biosynthesis, regulation, and function, Adv Enzymol Relat Areas Mol Biol 50 (1979) 177-242.
[4] S. Stockler, U. Holzbach, F. Hanefeld, I. Marquardt, G. Helms, M. Requart, W. Hanicke, J. Frahm, Creatine deficiency in the brain: a new, treatable inborn error of metabolism, Pediatr Res 36 (1994) 409-413.
[5] S. Stockler, D. Isbrandt, F. Hanefeld, B. Schmidt, K. von Figura, Guanidinoacetate methyltransferase deficiency: the first inborn error of creatine metabolism in man, Am J Hum Genet 58 (1996) 914-922.
[6] S. Stockler, F. Hanefeld, J. Frahm, Creatine replacement therapy in guanidinoacetate methyltransferase deficiency, a novel inborn error of metabolism, Lancet 348 (1996) 789-790.
[7] C.B. Item, S. Stockler-Ipsiroglu, C. Stromberger, A. Muhl, M.G. Alessandri, M.C. Bianchi, M. Tosetti, F. Fornai, G. Cioni, Arginine:glycine amidinotransferase deficiency: the third inborn error of creatine metabolism in humans, Am J Hum Genet 69 (2001) 1127-1133.
[8] G.S. Salomons, S.J. van Dooren, N.M. Verhoeven, K.M. Cecil, W.S. Ball, T.J. Degrauw, C. Jakobs, X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome, Am J Hum Genet 68 (2001) 1497-1500.
[9] E.H. Rosenberg, L.S. Almeida, T. Kleefstra, R.S. deGrauw, H.G. Yntema, N. Bahi, C. Moraine, H.H. Ropers, J.P. Fryns, T.J. deGrauw, C. Jakobs, G.S. Salomons, High prevalence of SLC6A8 deficiency in X-linked mental retardation, Am J Hum Genet 75 (2004) 97-105.