ALZHEIMER’S DISEASE: SORTING THROUGH TANGLES AND CLEARING UP THE PLAQUE…
In 1907, Dr. Alois Alzheimer published the first case study on a patient with what is now known as Alzheimer’s disease. Shakespeare almost beat him to it in the “All the World’s a Stage…” monologue from As You Like It, describing the last “scene” of a 7 part life ending in “second childishness and mere oblivion/Sans teeth, sans eyes, sans taste, sans everything.” The Canada Study on Health and aging estimated that 8% of the Canadian population over 65 years of age had a form of dementia and that 5.1% had Alzheimer’s disease (1). Alzheimer’s dementia has a devastating impact on affected individuals and their families and will become increasingly prevalent with the aging of the Baby Boomers. While there is currently no cure, an increasing understanding of disease pathology helps us to design potentially curable drugs.
What is dementia?
Dementia describes a long-term loss of memory, language and/or visual-spatial function in patients (2). Patients may also show mood and personality changes and may have trouble problem solving and concentrating. There are many diseases other than Alzheimer’s that are associated with dementia. Vascular dementia results from diseased or damaged blood vessels of the brain or from small blood clots which block blood flow to areas of the brain, depriving these areas of oxygen. Human immunodeficiency virus (HIV) can cause a form of dementia. Hypothyroidism, normal pressure hydrocephalus, and deficiencies or vitamins A, B1 or B12 can cause dementia that is reversible upon treatment. In the majority of other dementias, the underlying pathology is associated with clumps of proteins aggregating in areas of the brain. These include Huntington’s disease, frontotemporal dementia, Lewy Body dementia, and the prion diseases, in addition to Alzheimer’s disease.
How is Alzheimer’s a distinct form of dementia?
When you look at slices of brain of deceased Alzheimer’s patients under the microscope, there are two features that are distinct to this disease: plaques and tangles (Figure 1). When you mention plaque, most people think of visits to the dentist. Dental plaques are yellowish films made of complex sugars that help bacteria stick to your teeth. Alzheimer’s plaques are clumps of protein that aggregate in the space surrounding brain cells, or neurons. The protein found within these plaques is a shortened and misfolded form of amyloid precursor polypeptide (APP).
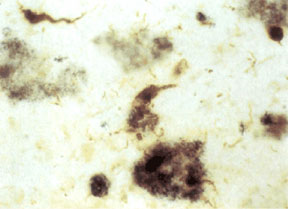
APP is a protein that spans cell membranes. It has many roles in neurons, including gene regulation, cellular cholesterol metabolism, transport of materials along neuronal axons and acting as a receptor (3). APP has several sites that are specifically cut by enzymes called secretases. α and β-secretases cut APP on its extracellular side of the membrane (4). γ-secretase cuts APP within the membrane (4). Different products are generated depending on whether α or β-secretase was the cutter. When α and γ-secretasescut APP, the product is soluble and plays no role in Alzheimer’s disease (4). The product cut out by β and γ-secretases is amyloidogenic (4). This means that a specific amino acid sequence within the peptide causes it to form β sheets instead of its normal helix shape. Multiple peptides stick to each other via their β sheets to form oligomers. It is the oligomers which are most toxic to neurons (5). However, much larger aggregates can form and these are the plaques we can see under electron microscopy and by special staining in brain slices.
Tangles in Alzheimer’s disease are quite literally tangled proteins. (6) The protein found in these tangles is called “tau.” In normal neurons, tau proteins are important in stabilizing the cell skeleton (7). They also transport molecules within the cells. Tau proteins in tangles are easy to recognize because they have many more phosphates attached than normal. Having too many phosphate groups attached may help tau proteins to form long polymers (8). Many polymers tangled together form a “tangle.” When tau accumulates in tangles it is no longer available to stabilize the cytoskeleton. As a result, neurons are fragile and prone to dying.
What is the pathogenesis of Alzheimer’s?
The brain can be simplified into two parts: the brain stem and the cortex. The brain stem controls breathing, heart rate, and many other involuntary functions that are essential for life. The cortex is involved in “higher functions” such as consciousness, memory, language abilities, and thought. It is the cortex which is affected in Alzheimer’s disease. Plaques and tangles are thought to play important roles in causing atrophy of the cortex. The more plaques and tangles found within a brain, the more atrophy and the more severe a patient’s dementia is (9). As the cortex atrophies, neurons and the synaptic connections between neurons are lost.
Another important feature of Alzheimer’s disease is the drastic changes in the quantity of neurotransmitters in the brain. Neurotransmitters are the chemical messengers which allow one neuron to talk to its neighour. One neuron releases a neurotransmitter from its tail and the neurotransmitter stimulates a receptor on the head of a second neuron. Neurotransmitters are usually modified amino acids or small proteins. In Alzheimer’s disease, the levels of seven important neurotransmitters are reduced in the cortex and the hippocampus (9). The reduction in the neurotransmitter acetylcholine (Ach) is the most severe, occurring most prominently in the basal forebrain. Cholinergic neurons are especially important in memory and their loss is attributed to worsening memory (9). Levels of the neurotransmitter glutamate are elevated in the brains of Alzheimer’s patients (10). Glutamate can overstimulate neurons to such an extent that is toxic and can kill neurons (9).
Is Alzheimer’s genetic?
There are three sets of the population with risks for developing Alzheimer’s. Down’s Syndrome patients commonly get early-onset Alzheimer’s disease (9). Secondly, a small number of people with a strong family history of early onset Alzheimer’s disease may have mutations in the presenilin-1 or presenilin-2 genes (9, 11). Alternately, they may have a mutation in the APP gene that alters how well a cleavage site fits one of the secretase enzymes (11). The other genetic risk factor is an e4 allele of the apolipoprotein E (apoE) gene (9). ApoE is a surface protein on complexes that transport cholesterol in the blood. It binds to a liver receptor to promote clearance of these complexes from the blood.
Do environmental factors play a role?
Many environmental agents have been proposed to promote the development of Alzheimer’s disease. Heavy metals were long thought to be important. High levels of aluminum are found in the brains of Alzheimer’s patients and it was hypothesized that aluminum promotes the formation of tangles (9). According to current research, aluminum exposure is not a major risk factor to Alzheimer’s disease (9). Cholesterol has recently been identified as an important risk factor (12). Significant contributing factors to high cholesterol include smoking and a diet high in saturated fat and cholesterol.
There are also protective factors against Alzheimer’s disease. Some studies have shown that people who take antioxidants, anti-inflammatory drugs, statins, or estrogen over the long-term are partially protected against Alzheimer’s (13, 14). Intellectual stimulation, university education, physical exercise and social interactions have all been reported to have positive effects in preventing the disease (15, 16).
How do we diagnose and follow the progress of the disease?
We currently diagnose Alzheimer’s disease based on behavioral symptoms. These include aphasia, agnosia, apraxia, depression, hallucinations, emotional and physical outbursts, insomnia, illusions, incontinence, and abnormal muscle tone (9). Special tests such as the Mini-mental standard exam may be used to assess cognitive and memory impairment. Brain atrophy can be shown by a CT or MRI scan (9).
To confirm a diagnosis of Alzheimer’s, we would need to biopsy the brain. This is not done. However, after a patient’s death a pathologist usually confirms the diagnosis by analysis of brain tissue. Several techniques are being developed to diagnose and monitor the disease by MRI and PET imaging. A patient would be given a compound that binds to any amyloid plaques in the brain and accumulations of the compound in the brain would be picked up by the imaging machine (17). This would allow us to confirm diagnoses while a patient was alive and would allow us to track the progression of the disease.
What are current therapies for Alzheimer’s?
Currently, there is no cure for Alzheimer’s disease. In patients with mild Alzheimer’s, rivastigmine, donepezil, or galantamine are given to slow the breakdown of Ach by enzymes (9, 18). The theory behind these drugs is that by making any Ach released by neurons stay around longer, it can compensate for the “cholinergic deficit” that partially explains memory loss in Alzheimer’s. However, the effectiveness of these drugs is limited. Patients with more advanced disease may also be given memantine (9, 18). This drug prevents glutamate from binding to its receptors and therefore should limit how toxic glutamate is to neurons. Atypical antipsychotic drugs may be given to control behavioral symptoms (9). Many patients are also prescribed antioxidants, specifically vitamin E and selegiline, and the herbal remedy ginko biloba (19).
How do we study Alzheimer’s disease?
To develop a cure for a disease, it is important to have a disease model. Due to ethics, human studies are very limited. Instead, scientists have attempted to create models of Alzheimer’s disease in mice. Mice do not naturally develop Alzheimer’s. However, as mice and humans age, they both develop mild cholinergic deficits. Initial studies focused upon understanding the development of cholinergic deficits and attempting to minimize this development (20). Later studies of cholinergic deficits used the drug scopolamine on mice. Scopolamine is a drug which blocks Ach from acting on its receptors in the brain. Models of glutamate glucotoxicity have also been used to study how to minimize this process in Alzheimer’s (20). Other early models involved making lesions in areas of the mouse’s brain that normally atrophy in Alzheimer’s disease. These models allowed us to better understand how atrophy of specific brain regions contributes to specific symptoms of Alzheimer’s disease. A final model involving naturally-occuring mice is that of the SAMP8 mouse. This strain of mouse ages quickly and near the end of its life, it develops a small degree of plaque deposition in its brain.
More powerful models of Alzheimer’s disease involve transgenic mice (20). A transgenic mouse has had one or several genes from another species of organism introduced into its genome. For example, to induce amyloid development in mouse brains the human APP gene was inserted into the mouse genome. Another gene introduced to induce amyloid formation in mice is PSEN, a gene involved in the cleavage of APP to amyloid beta. Mice which have both the human APP and PSEN genes have enhanced amyloid deposition and are even stronger models of disease pathogenesis. Transgenic mice have also been developed to study tau pathology. These mice express the human tau gene and develop tangles. The limitations of APP, APP/PSEN and tau transgenic mice is that they only develop either plaques or tangles. As both processes may be important in the disease process double (APP/tau) and triple (APP/PSEN/tau) transgenic mice have been developed. When a scientist chooses a mouse model to study a particular question about the disease process, he will choose the most relevant one. For example, to study whether an anti-amyloid drug is effective an APP or APP/PSEN mouse may be sufficient to see if the drug can prevent plaques from forming. The complex double and triple transgenic mice are more useful to answer complicated questions relevant to development of the disease.
What are the directions of new potential therapies?
A cure for Alzheimer’s would be a goldmine for any pharmaceutical company. As such, many approaches have taken to seek such a cure. Some approaches focus on the immune system. As the incidence of Alzheimer’s is lower in people taking anti-inflammatory drugs over the long-term for other conditions, approaches to limit inflammation may slow disease pathogenesis (21). Immunotherapy, in which a vaccine teaches the immune system to remove amyloid from sites where it is accumulating, was shown to be promising in mouse models. However, human trials were halted due to serious complications (22). Other approaches focus upon halting amyloidopathy or tauopathy. One approach to prevent amyloid accumulation is to minimize the amount of APP broken down to amyloid beta using beta and gamma secretase inhibitors (18). Alternatively, compounds which directly inhibit amyloid-beta peptides from forming amyloid oligomers and fibrils are being developed (18). Three approaches are being used to prevent tangle formation. Tau kinase inhibitors would prevent the hyperphosphorylation of tau proteins, while tau phoshatase activators would promote removing phosphate groups from hyperphosphorylated tau molecules (18). Tau aggregation inhibitors would prevent the polymerization of tau proteins (18). The newest approach, control of cholesterol metabolism, stems from recent evidence showing a strong correlation between high blood cholesterol and the development of Alzheimer’s disease (23).
References
1) [No authors listed] 1994. Canadian Medical Association Journal. 150(6):988-913.
2) Goldman L and Ausiello D. Cecil essentials of medicine. 22nd Ed. W.B. Saunders Company, 2004.
3) Turner PR, O’Connor K, Tate WP and Abraham WC. 2003. Progress in Neurobiology. 70(1):1-32.
4) Pearson HA and Peers C. 2006. Journal of Physiology. 575(1):5-10.
5) Walsh DM, Klubyin I, Fadeeva JV, Rowan MJ and Selkoe DJ. 2002. Biochemical Society Transactions. 30(4):552-7.
6) del C. Alonso A, Zaidi T, Novak M, Grundke-Iqbal I and Iqbal K. 2001. Proceedings of the National Academy of Science. 98(12):6923-8.
7) Alberts B, Bray D, Lewis J, Raff M and Roberts K. Molecular Biology of the Cell. 2nd Ed. Garland Publishing, 1989.
8) Goedert M and Spillantini MG. 2006. Science. 314(5800):777-781.
9) Goetz CG. Textbook of Clinical Neurology. 2nd Ed. Saunders, 2003.
10) Koch HJ, Uvanik G and Fischer-Barnicol D. 2005. Current Drug Targets. CNS and neurological disorders. 4(5):499-506.
11) Schutte DL. 2006. American Journal of Nursing. 106(12):40-7.
12) Canevari L and Clark JB. 2006. Neurochemical Research. [Epub ahead of print]
13) Scherger JE, Misulis K, Murray JL and Galloway GM. First Consult, MD Consult Website, Elsevier, 2007.
14) Wolozin B and Bednar MM. 2006. Neurological Research. 28(6):630-6.
15) Fratiglioni L, Paillard-Borg S and Winblad B. 2004. Lancet Neurology. 3(6):343-53.
16) Roe CM, Xiong C, Miller JP and Morris JC. 2007. Neurology. 68(3):223-8.
17) Wu W and Small SA. 2006. Curr Alzheimer Res. 3(5):529-39.
18) Klafki HW, Staufenbiel M, Kornhuber J and Wiltfang J. 2006. Brain. 129(11):2840-55.
19) DeLaGarza VW. 2003. Am Fam Physician. 68(7):1365-72.
20) Van Dam D and De Deyn PP. 2006. Nature Reviews Drug Discovery. 5:956-70.
21) Wyss-Coray T. 2006. Nature Medicine. 12(9):1005-15.
22) Wang YJ, Zhou HD and Zhou XF. 2006. Drug Discovery Today. 11(19-20):931-8.
23) Whitfield JF. 2006. Expert Opin Investig Drugs. 15(12):1479-85.