METAGENOMIC METHODS REVEAL MARVELOUS MICROBIAL MICROCOSMS
Do you know how many different kinds of microbes exist on Earth? It may sound like a silly question but please indulge me and try to come up with a number… ready for the answer…? I hate to disappoint you but… it turns out that we still do not know! For most of the twenty-first century, few biologists contested the view that plants and animals accounted for the majority of the planet’s diversity (Curtis et al. 2006). Their perspective on microbial diversity was deeply skewed by the fact that they only recognized the select few species capable of growing under artificial conditions in a lab. Furthermore, with few obvious physical characteristics to differentiate between microbial groups, microscope analysis yielded only limited information about their diversity (Fierer & Lennon 2011). Only in recent years have microbiologists (those who study microbes) learned that they had grossly underestimated microbial diversity: we now know that over 99% of microbial species observed in nature cannot be cultivated in a lab (presumably because they have requirements that cannot be recreated), and microbes actually comprise the majority of species on our planet (Hugenholtz et al. 1998, Malik et al. 2007). By now you must be wondering: what caused such a radical change in our understanding of microbial diversity? And perhaps more importantly: why the heck should we care?
“Microbe” is a generic term used to describe a wide variety of organisms that are invisible to the naked eye (microscopic) and usually consist of a single cell (unicellular). Types of microbes include tiny algae and fungi, bacteria, and archaea (a diverse and abundant group of unicellular organisms found in most environments but discovered only about 35 years ago) (Gribaldo & Brochier-Armanet 2006). Microbes are found everywhere: from oceans, lakes, soil, and the guts of animals, to more exotic locations like the troposphere, underwater volcanoes, nuclear reactors, and the polar deserts of Antarctica (Freeman et al. 2011; Fierer & Lennon 2011). Microbes are also very abundant; imagine that a teaspoon of good-quality soil contains billions of microbial cells (Freeman et al. 2011). It is no exaggeration to say that microbes dictate life on Earth; they drive the geochemical cycles that convert essential elements such as nitrogen, carbon, and phosphorous into a form that is usable by organisms in aquatic and terrestrial ecosystems. Through their intimate associations with plants and animals, microbes facilitate nutrient acquisition and absorption, but they are just as capable of causing disease. Microbiologists are just now beginning to understand the connection between some microbe’s biological properties and the role they play in nature, and much of this new knowledge has been obtained by looking at the microbes’ genomes.
What is a genome anyway? In a nutshell, it is the total DNA of an organism. You can think of DNA as containing the blueprint (in the form of genes) for making the products (such as proteins and enzymes) that give an organism its specific characteristics. So a genome is like a library of DNA that is passed from parent to offspring and contains all the necessary information to build, operate, and maintain an organism. All species have a distinctive genome, which is why humans have a pair of eyes, arms, and legs, but no wings or horns – these traits are encoded in our genomes. Using fancy machines and computers, scientists in the field of genomics can read (or sequence, in scientific speak) and interpret the whole genome of an organism (Steward & Rappe 2007). Unprecedented advances in biotechnology and computational capacity in the last decade have made genomic tools much cheaper, faster, and capable of sequencing the genomes of many organisms simultaneously. The newest technique is aptly called “next-generation sequencing” (NGS), and it has made the sequencing of one unit of DNA 100 million times cheaper and 30,000 times faster than in 1990 (Resnick 2011). This means that biologists can now go nuts and sequence as many genomes as their little hearts desire, and it is precisely this phenomenon that gave rise to the emerging field of metagenomics. Whereas genomics was concerned with the genome of a single individual, “meta” describes the use of genomic tools on all the organisms that share a common environment. The collective genome of a community of microbes is referred to as the metagenome. To date, the metagenomic approach has been applied exclusively to microbial communities using DNA obtained directly from an environmental sample such as a cup of seawater, a piece of rock, or a swab of a person’s belly button! Thus, metagenomic tools allow microbiologists to bypass the need to cultivate microbes in a lab to learn about them, effectively widening the “lab cultivation bottleneck” that limited knowledge about the ecological diversity and function of microbes. Although still in its infancy and with important kinks to iron out, the metagenomic method has already generated impressive findings, such as never-before-seen genes and proteins (Hugenholtz & Tyson 2008). But rather than going into the nuts and bolts of the metagenomic method, I would like to share with you two examples that showcase the potential for it to revolutionize human health, and contribute to the resolution of one of the world’s most pressing problems: pollution (Fraser-Liggett & Weissenbach 2007).
Humans are a walking and talking habitat with bacteria and archaea blanketing every surface. Microbial cells outnumber human cells ten to one, and the vast majority of them inhabit our gut (Sleator et al. 2008; Gill et al. 2006). Microbes in our guts (our gut microbiota) do the jobs that our bodies have not evolved to do on their own, such as obtaining nutrients that would otherwise be inaccessible, making essential vitamins, degrading complex sugars, and contributing to the development of our immunue system (Sleator et al. 2008; Kurokawa et al. 2007). The diagnosis of gastrointestinal diseases currently focuses on a single type of microbe that is either ruled in or out as the disease-causing agent after being isolated and grown in the lab. Although this method is perfectly appropriate for detecting and treating certain illnesses, we now know that our guts host a diverse community of microbes with complex interactions. A metagenomic survey of the human gut found nearly 40,000 bacterial species, most of which had never been cultivated in a lab, and concluded that large-scale microbial imbalances in our digestive tract, and not single organisms, are associated with a diversity of conditions like obesity, Crohn’s disease, and antibiotic-associated diarrhea (Frank & Pace 2008; Manichanh et al. 2006). Most metagenomic studies of the human gut have used a “sequence-driven” approach, obtaining a metagenome by cutting up all the DNA from an environmental sample into small fragments that are then sequenced. Researchers find which genes are present in their metagenome by comparing their sequenced DNA fragments with those in a database that contains all the microbe genes that have ever been found (Figure 1). The next step, and one that is still a huge challenge, is to understand how the collective genes influence the function of microbial communities, and by extension the health of the person that hosts them (Frank & Pace 2008). This was done successfully by Turnbaugh et al. (2007): by looking at the gene content of gut microbial communities of obese mice, they predicted a microbiota with an increased capacity to convert indigestible sugars into compounds that are absorbed and stored as fat in the mice. They followed-up with an experiment in which mice with normal weights were colonized with microbiota from either obese mice or lean mice. Remarkably, the mice that received “obese microbiota” had significantly more fat accumulation than those with “lean microbiota”, confirming the initial prediction but also showing that microbiota that increase susceptibility to obesity can be transferred from one host to another.
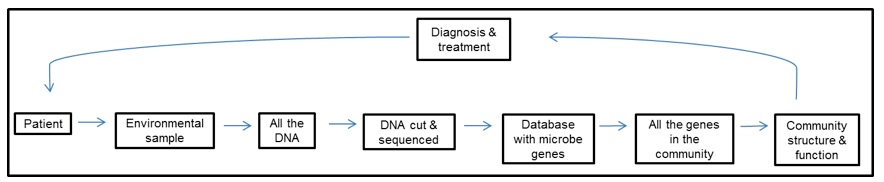
Figure 1. [Click on image to enlarge] Sequence-based approach for studying human metagenomes. (Adapted from Frank & Pace 2008).
By using metagenomics, researchers can approach microbial communities in humans as complex ecosystems whose structure, dynamics, and functions influence the health of their host (Frank & Pace 2008). The Human Microbiome Project (HMP) hopes to push this approach even further by designing and implementing strategies to manipulate the human microbiota to optimize its performance in the context of an individual person’s health (Turnbaugh et al. 2007). To this purpose the HMP is using metagenomics to study microbial communities of the human gut, nose, skin, mouth, and genitals (HMP 2012). But before metagenomics can be used as a tool in personalized medicine, a set of reference metagenomes from healthy individuals must be established for all microbiota of interest. This will serve as a genetic guideline for a healthy microbiota that can be used as a point of comparison for future metagenomic samples and will allow scientists to discern disease-causing variations. A reference metagenome would have to be representative, and therefore assembled by pooling together data obtained from several individuals. It is still unclear how different microbiota vary with age, gender, ethnicity, diet, environment (urban vs. rural), and lifestyle. Thus, sampling to establish reference metagenomes will be a long and challenging task, but one that promises rewarding outcomes (Turnbaugh et al. 2007).
Microbial abundance in aquatic and terrestrial environments is just as impressive as it is in humans – it was recently estimated that 1016 microbes are present in one ton of soil, while a mere 1011 stars exist in the entire galaxy (Desai et al. 2010). Microbes are the invisible, tireless, and thankless workers that maintain ecosystem productivity and health by cycling essential nutrients, and in some cases by breaking down harmful compounds. As the world population increases, so does the amount of pollutants released into the environment, be it as byproducts of every day life or as accidental leaks and spills (Desai et al. 2010). This has led to the widespread presence of xenobiotics, which are foreign chemicals that are found in an organism but were not produced by the organism itself (Freeman et al. 2011). Many xenobiotics are not easily degraded by the enzymes present in an ecosystem, which results in their accumulation in soils, water, and living organisms. To make matters worse, there is evidence that some xenobiotics are toxic, cause mutations, and lead to cancer in many organisms (Eyers et al. 2004). Amazingly, some microbial communities have no problems establishing and thriving in contaminated sites. Instead of being poisoned by pollutants, certain species of microbes can harness them to carry out internal functions and funnel them back into the environment after rendering them nontoxic (Eyers et al. 2004; Freeman et al. 2008; Desai et al. 2010). Although scientists have known about these microbe superpowers for some time, the biological mechanisms behind a microbe’s ability to adapt to contaminated environments remain poorly understood.
The conditions of contaminated sites are difficult to mimic in a lab, so the diversity and genetic properties of microbes that break down pollutants goes undetected in studies that rely on lab cultivation because many species do not survive (Malik et al. 2008). Thus, metagenomic sampling in contaminated sites is allowing researchers to access the genes, proteins, and enzymes of microbial communities capable of breaking down contaminants, and this is providing valuable knowledge for the field of bioremediation. Bioremediation is defined as the use of living organisms, usually bacteria and archaea, to degrade environmental pollutants (Freeman et al. 2011). It is considered an eco-friendly and cost-effective way of restoring health in polluted ecosystems (Desai et al. 2010). Bioremediation can be ex-situ, by removing contaminants from a site and remediating them in a separate facility. This method relies on microbes that can be cultivated artificially and mass produced. With in-situ bioremediation, pollutants are remediated on-site by introducing foreign microbes or by stimulating naturally-occurring microbes capable of breaking down contaminants (Desai et al. 2010). Only in-situ bioremediation is feasible in remote areas, and there is a sense of urgency in developing such strategies in the face of climate change, which will increase interest and traffic in areas like the Arctic for research and resource exploration. Experiments in the Arctic have shown that native microbes can bioremediate xenobiotics introduced by diesel spills. However, bioremediation efficiency in different study sites was mixed: sites where the soil was excavated had the highest efficiency, but until recently the biological reason for this was unknown. Yergeau et al. (2012) used a “sequence-driven” metagenomic approach to sequence the metagenome of contaminated Arctic soil with the goal of finding the types of microbes and genes present in the sites with the highest bioremediation rates. By comparing the metagenomes sequenced from contaminated soil samples to those from uncontaminated samples, the researchers found major differences in microbial community structure. Four microbe groups that need oxygen to live dominated the contaminated metagenomes, and they are all associated with genes for breaking down xenobiotics. Excavating soils supplies them with oxygen, which explains why the contaminated sites that were excavated had the highest rates of xenobiotic break down. The same four microbe groups were present in uncontaminated soil, but in smaller numbers, showing that these microbes can take advantage of the presence of diesel xenobiotics to thrive. Although the findings of this study may not be applicable to any other ecosystem, it illustrates how metagenomics can be used to design bioremediation strategies that specifically target microbes with the highest remediation capacity.
The amount of an organism’s genome that is recovered from an environmental sample depends on the relative abundance of that organism. A limitation of today’s metagenomic tools is that they are not sensitive enough to pick up on rare genomes, which could result in studies missing organisms that, in spite of their low abundance, have important ecological functions. Another limitation is the difficulty of matching anonymous DNA fragments to their owners, for which long DNA fragments are necessary, but not easy to obtain with today’s tools (Hugenholtz & Tyson 2008). In spite of its current shortcomings, it is undeniable that metagenomics has triggered an age of exploration and discovery in the microbial world. The fields of biology, medicine, and ecological health are undergoing transformations in the way microbial communities are studied. And even though our knowledge about microbes is being amassed at an unprecedented rate, we are still far from understanding the full extent of microbial biodiversity, genetic properties, and community interactions (Hugenholtz & Tyson 2008). The way I see it, and what I have hopefully conveyed here, is that the study of microbes is not only an inherently fascinating topic, but also one with profound practical applications.
REFERENCES
Curtis, T. P, I. M. Head, M. Lunn, S. Woodcock, P. D. Schloss, W. T. Sloan. 2006. What is the extent of prokaryotic diversity? Phil. Trans. R. Soc. B. 361, 2023-2037.
Desai, C., H. Pathak, D. Madamwar. 2010. Advances in molecular and “-omics” technologies to gauge microbial communities and bioremediation at xenobiotic/anthrophogen contaminated sites. Bioresource Technology 101, 1558-1569
Eyers, L., G. L. Schuler, B. Stenuit, S. N. Agathos, S. El Fantroussi. 2004. Environmental genomics: exploring the unmined richness of microbes to degrade xenobiotics. Appl Microbiol Biotechnol 66: 123-130.
Fierer, N. and J. T. Lennon. 2011. The generation and maintenance of diversity in microbial communities. Am. J. Bot. 98(3): 439-448.
Frank, D. N. and N. R. Pace. 2008. Gastrointestinal microbiology enters the metagenomics era. Current Opinion in Gastroenterology. 24(1): 4-10.
Fraser-Liggett, C.M. and J. Weissenback. 2007. Genomics. Editorial Overview. Current Opinion in Microbiology, 10: 479-480.
Freeman, S., M. Harrington, J. Sharp. Biological Science. Volume 1. Toronto, Ontario: Benjamin Cummings.
Gill, S. R., M. Pop, R. T. DeBoy, P. B. Eckburg, P. J. Turnbaugh, B. S. Samuel, J. I. Gordon, D. A. Relman, C. M. Fraser-Liggett, K. E. Nelson. 2006. Metagenomic analysis of the human distal gut microbiome. Science. 312.
Gribaldo, S. and C. Brochier-Armanet. 2006. The origin and evolution of Archaea: a state of the art. Phil. Trans. R. B. 361, 1007-1022.
Hugenholtz, P., B. M. Goebel, N. R. Pace. 1998. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180(18): 4765
Hugenholtz, P. and G. W. Tyson. 2008. Metagenomics. Nature Q & A, 455.
Human microbiome project. http://commonfund.nih.gov/hmp/ Accessed: 05/11/12.
Kurokawa, K., T. Itoh, T. Kuwahara, K. Oshima, H. Toh, A. Toyoda, H. Takami, H. Morita, V. K. Sharma, T. P. Srivastava, T. D. Taylor, H. Noguchi, H. Mori, Y. Ogura, D. S. Ehrlich, K. Itoh, T. Takagi, Y. Sakaki, T. Hayashi, M. Hattori. 2007. Comparative metagenomics revleaed commonly enriched gene sets in human gut microbiomes. DNA Res. 14(4): 169-181.
Malik, S., M. Beer, M. Megharaj, R. Naidu. 2008. The use of molecular techniques to characterize the microbial communities in contaminated soil and water. Environmental International 34, 265-276.
Manichanh, C. L. Rigottier-Gois, E. Bonnaud. 2006. Reduced diversity of faecal microbiota in Crohn’s disease revealed by metagenomic approach. Gut. 55:205-211.
Resnick, R. 2011. Richard Resnick: Welcome to the genomic revolution [Video file]. Retrieved from http://www.ted.com/talks/richard_resnick_welcome_ to_the_genomic_ revolution.html
Sleator, R. D., C. Shortall, C. Hill. 2008. Metagenomics. The Society for Applied Microbiology. Letters in applied microbiology 47, 361-366.
Steward, G. F., and M. S. Rappe. 2007. What’s the ‘meta’ with metagenomics? The ISME Journal, 1, 100-102.
Turnbaugh, P. J., R. E. Ley, M. Hamady, C. M. Fraser-Liggett, R. Knight, J. I. Gordon. 2007. The human microbiome project. Nature Insight feature. 449.
Yergeau, E., S. Sanschagrin, D. Beaumier, C. W. Greer. 2012. Metagenomic analysis of the bioremediation of diesel-contaminated Canadian high Arctic soils. PLoS ONE 7(1): e30058.