GENOME PROJECTS: UNCOVERING THE BLUEPRINTS OF BIOLOGY
(August 2003)
In the early days of genetics, scientists did not have the resources to look at more than a few genes at a time. This made the process of understanding the influence of genetics on an organism slow and arduous. Scientists were faced with the enormous task of attempting to understand genetic influence with little information to complete the task. The understanding of genes would have been very helpful in solving this problem.
The year 1995 saw the completion of the first two complete non-viral genomes, Haemophilus influenzae [1] and Mycoplasma genitalium [1], two bacteria that can cause human disease. Since then, over 100 genomes have been fully sequenced, including those of higher organisms like baker’s yeast, the fruit fly, and the nematode [2]. With the announcement in June of 2001 that the first draft of the human genome had been completed [3], scientists’ approach to biology completely changed. The entire set of human genes was now available. This represented an irresistible amount of data that breached the bioinformatic gap that lay between biologists and their understanding of genetics.
To begin to see the significance of such an historical event, it is necessary to look at why uncovering a genome is an important biological task.
What is a Genome?
The genome refers to all DNA present in an organism.
DNA is the “genetic blueprint” that determines the genotypic make-up of each organism. In its barest form, DNA consists of two strings of nucleotides, or bases (abbreviated A, C, G, and T), wound around each other. The bases composing DNA have specific binding capabilities: A always binds to T, and C always binds to G. These binding capabilities are useful for scientists to understand since, if the nucleotide sequence of one DNA strand is determined, complementary binding allows the sequence of other strand to be deduced.
In the case of humans, DNA is organized into 24 structural units called chromosomes. Each chromosome consists of compacted coils of DNA. While much of this DNA has no known function (these stretches of DNA are conveniently referred to as spacer DNA or junk DNA), a significant portion of the DNA codes for genes. Each gene provides the information necessary to produce a protein, which is responsible for carrying out cellular functions. The complement of proteins in an organism is very important, with diseases often manifesting when a protein does not function properly.
Why Sequence Genomes, Especially Non-Human Genomes?
One of the interesting things about biological organisms is their remarkable similarity at the molecular level, despite their obvious outward differences. For instance, many genes are found in morphologically different organisms despite the phylogenetic distance between them4. Not only are these genes very similar in their DNA sequence composition; they also tend to perform the same functions. Thus, by understanding the function of a gene in one organism, scientists can get an idea of what function that gene may perform in a more complex organism such as humans. The knowledge gained can then be applied to various fields such as medicine, biological engineering and forensics.
The Sequencing Reaction: How the Nucleotide Composition of DNA is Determined
To understand how DNA is sequenced, one must first know a little about the structure of DNA:
- A segment of DNA, which is ordinarily double stranded, has a specific orientation, as it has a 5′ (read as “5 prime”) and a 3′ (“3 prime”) end. This can be simply thought of as a front and tail end to the DNA segment.
- When DNA is synthesized in the lab, the two strands are separated and new bases are added to the 3′ end-thus DNA is assembled from the 5′ to 3′ end.
- DNA cannot be synthesized from scratch. A short piece of DNA, called a primer, is required for the reaction to begin.
- Primers are designed such that they are able to bind to the target DNA, the binding of which is the initiator for DNA synthesis.
DNA sequencing is accomplished by the Fredrick Sanger method (see Figure 1), for which he won his second Nobel Prize in 1980.
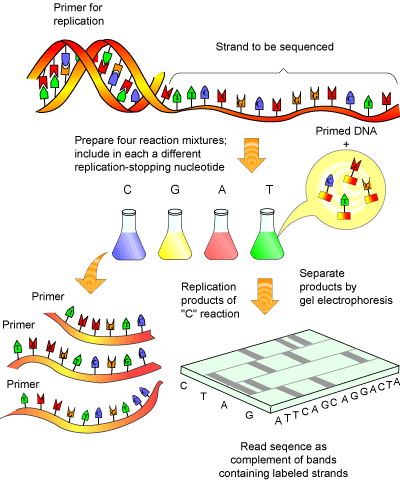
This method essentially involves amplifying a single stranded piece of DNA many times [5]. Normally, when DNA is amplified, new deoxy-nucleotides ( dNTPs) are added as the strand of DNA grows. The Sanger method employs special bases called dideoxy-nucleotides ( ddNTPs). These are similar to dNTPs, except for two important differences: they have fluorescent tags attached to them (a different tag for each of the 4 ddNTPs) and are missing a crucial atom that prevents new bases from being added to a DNA strand after a ddNTPs has been added. Thus, once a ddNTP is inserted into a growing DNA strand, synthesis of that strand is stopped. After many repeated cycles of amplification this will result in all the possible lengths of DNA being represented and every piece of synthesized DNA containing a fluorescent label at its terminus.
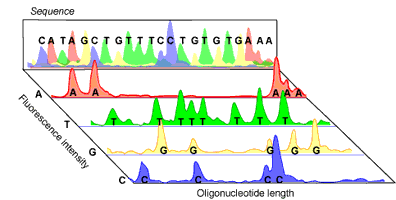
Amplified DNA can then be separated according to size via gel electrophoresis. As the fluorescent DNA reaches the bottom of the gel (now separated from smallest to largest), a laser can pick up the fluorescence of each piece of DNA. The trick to the Sanger method lies in the fact that each ddNTP emits a different fluorescent signal, so that the presence of a ddNTP at the terminus can be recorded on a computer (see Figure 2). The reaction is set up so that a fluorescent ddNTP is present at every position in the DNA strand (i.e. every possible size of DNA strand is present) so that every nucleotide in the strand can be determined. A computer program can then compile the data into a coloured graph showing the determined sequence.
In the past, the separation of the DNA strands by electrophoresis was a time consuming step, requiring the use of radioisotopes for labelling ddNTPs. This was less than trivial, as four different sequencing reactions were required (one for each ddNTP) and the resulting sequencing gel needed to be analyzed manually. Today, fluorescent labels and new advances in gel electrophoresis have made DNA sequencing not only fast and far more accurate, but also almost fully automated, including the read out of the final sequence.
Sequencing a genome
While the Sanger method is the accepted method for sequencing DNA, one cannot sequence a complete genome using this method alone. The main reason for this is that as the pieces of DNA get larger, resolving two pieces by one base becomes virtually impossible [6]. In fact, only about 1000 bases can be sequenced accurately, a far cry from the 50 to 250 million bases that comprise a human chromosome. Furthermore, as stated above, a primer of known sequence is required for each sequencing reaction. Thus, one cannot take any piece of DNA and “just sequence it.” A known starting point, and thus some knowledge of the sequence, is required to begin the reaction. To circumvent this problem, DNA is usually cut up into smaller, more manageable chunks and then placed into a small circular piece of DNA known as a plasmid or cloning vector (a process generally referred to as cloning). The cloning vector’s sequence is known and therefore allows any piece of DNA introduced into it to be sequenced.
With these ideas in mind, scientists set out to design methods to make possible the sequencing of an entire genome. No small task when you consider that the human genome contains approximately three billion bases that needed to be sequenced.
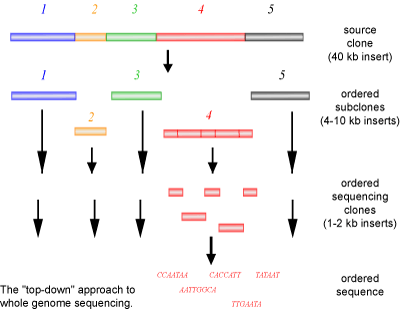
The first method of sequencing a genome, employed by the publicly funded Human Genome Project, involves cloning a large piece of DNA into smaller pieces called sub-clones. With the use of known genetic markers (i.e. physical characteristics that have been attributed to specific areas of a chromosome) a simple and poorly resolved map of where the sub-clones would be located on a chromosome is prepared. This allows the sub-clones to be placed in an order based on the structure of the chromosome. Each individual sub-clone is then sequenced. The resulting sequence is used to create a new primer to sequence flanking regions of the DNA that could not be sequenced in the first round of reactions. This process is continued until the sequences overlap (are contiguous). These contiguous sequences can then be assembled into a group of overlapping sequences, termed a contig. As this method progresses, larger and larger contigs will be produced, until a single ordered contig of the genome is achieved.
A common named for the above method is a ‘top-down’ approach (See Figure 3). If you look at a jigsaw puzzle as an analogy, a top-down approach is similar to starting the puzzle form one corner and working your way down and across in an ordered manner, always building on the last piece that was added. The advantages of this method are that each individual clone can be sent to different people for sequencing and that each stretch of DNA only needs to be sequenced once, as the DNA has already been mapped. However, a large disadvantage to this method is the slow process of sub-cloning and mapping of the clones, requiring significant human manipulation.
A second method is the so-called ‘shotgun’ method of sequencing (see Figure 4), which was employed by the privately funded company Celera Genomics to sequence the human genome. This method was the subject of a good deal of debate, as it is relatively crude in comparison to the method employed by the Human Genome Project. It involves each contig being sub-cloned into smaller fragments in the same way as the top-down approach, with the exception that a physical genetic map is not created. Instead, each clone is sequenced first, and then overlapping sequences are joined together to create the contig. In other words, random clones are sequenced (as they are not ordered) in the hopes that overlapping sequences will be found to piece together the contiguous sequence.
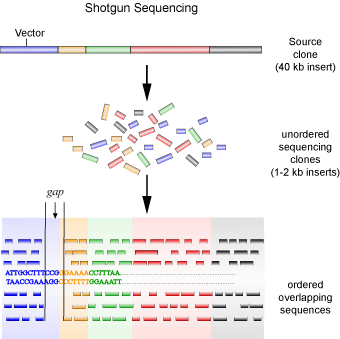
Using the jigsaw puzzle analogy again, the shotgun method is similar to starting with random pieces of the puzzle and looking for pieces that fit to it, regardless of where in the puzzle the piece originated from. One major problem with this method is uncertainty. You lack an initial map to guide you, making it difficult to be sure that the entire contig is represented. To get around this problem, the same contig needs to be sequenced many times to ensure that the probability of missing a sub-clone is less than 1%. After which the gaps between contigs must still be filled in, usually through the use of a technique called chromosome walking. The shotgun method is advantageous in that the laborious process of mapping and sub-cloning, requiring human hands, is eliminated. So, while this method requires much more sequencing compared to the first, it proves to be much more economical and faster due to the sequencing reactions being virtually fully automated and the sequences being assembled by computer programs.
When is a Genome Sequence Finished?
When it was announced that the first draft of the human genome was completed [3], it was commonly misreported by many media outlets that the human genome was sequenced. In fact, much more sequencing needs to be done to finish the job. This is because the genome sequence was still in the ‘draft’ stage, meaning that the genome had been sequenced about 4 to 5 times, and the data organized into fragments that are approximately 10,000 bases in size.
To prepare a high quality sequence of the human genome, potential errors in the sequence must still be statistically removed. This is done primarily by closing the gaps between contigs with additional sequencing, ultimately reducing ambiguity and ensuring that there is at most 1 error in every 10,000 bases. The finished version will require that a chromosome be sequenced about 9 to 10 times. Furthermore, not all regions of the chromosome can be cloned, resulting in them being unavailable for sequencing. Luckily, these regions, called heterochromatin, consist of telomeres and centromeres (the tips and centre of the chromosome, respectively), which are rich in repeating sequences (making cloning very difficult) and low in genes. Most of the genes reside in euchromatin, the part of the chromosome that can be sequenced. Therefore, a complete genome sequence actually refers to a high quality sequence of an organism’s euchromatin.
Benefits of Sequencing Projects
Why do we want to determine the A’s, T’s, C’s, and G’s of an organism?
When you get right down to it, a genome is the blueprint of how an organisms functions. If we are interested in understanding the complexity of life (and every biologist and doctor is), having a genome to study is a big step forward.
Scientists are revving up their computers to study genomes and the benefits of this are already being seen. Take the field of medicine as an example. As the population begins to become increasingly health conscious, more attention is being paid to the ongoing research in the medical sciences. As the chromosome maps have become more detailed, genes associated with genetic diseases such as Alzheimer’s disease [7] and familial breast cancer [8] have been identified. This has led to the hope that these diseases can be identified early and that new drugs and treatments can be discovered.
Genome projects also give us insight into other organisms, which has many applications in the industrial sector [9]. Increasing knowledge about domesticated plants and animals can reduce costs in agriculture, for example, by reducing the need for pesticides. Microbes are also an important resource. It has already been shown that bacteria can be used to clean up toxic chemical and oil spills and aid in the clean-up of sewage and waste. Bacteria have also been used to replace many industrial processes that require large amounts of toxic reagents or harsh conditions, making many workplaces, and their surrounding environment, much safer.
Final Words: Where is Genome Science Taking Us?
Even though the numbers of completed genomes is ever increasing, the real work is just beginning. New advances in technology must accommodate the increasing amount of data, as the information available to researchers can be overwhelming. Already new fields of science have been created by the sequencing of genomes. An example of this is functional genomics, which aims to look at the practical aspects of sequenced genomes by looking at genome-wide responses to various elements.
Finally, a whole can of ethical issues have been opened as researchers have begun patenting genes in the hopes of financial reward. Is it right to patent genes that are present in all humans? Who controls the genetic information? Can the use of genetic information oppress and control people, like in the movie Gattaca? Only education, debate and time will produce these answers.
Texts Consulted and Additional Reading
1. Dale JW, von Schantz M. 2002. From Genes to Genomes: Concepts and Applications of DNA Technology. West Sussex, England / New York: Wiley. 360p.
2. Town C, ed. 2002. Functional Genomics. Dordrecht/Boston: Kluwer Academic. 200p.
3. Caporale LH. 2003. Darwin in the Genome: Molecular Strategies in Biological Evolution. New York: McGraw-Hill. 245p.
4. Rangel P, Giovannetti J. 2002. Genomes and databases on the Internet: A Practical Guide to Functions and Applications. Wymondham: Horizon Scientific. 223p.
5. Primrose SB, Twyman RM. 2003. Principles of genome analysis and genomics. Malden, MA: Blackwell Pub. 263p.
References
1. Two Bacterial Genomes Sequenced. 1995. Human Genome News, May-June 7(1).
2. Genome-Scale Science. National Centre for Biotechnology Information:
3. The Genome International Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921.
4. Griffiths et al, eds. 2002. Modern Genetic Analysis: Integrating Genes and Genomes. New York: W.H. Freeman and Co. 736p.
5. An animation of the sequencing reaction from the Dolan DNA Learning Center.
6. Alphey L. 1997. DNA Sequencing: From Experimental Methods to Bioinformatics. New York: Springer. 206p.
7. Lahiri DK, et al. 2003. A Critical Analysis of New Molecular Targets and Strategies for Drug Developments in Alzheimer’s Disease. Curr Drug Targets 4(2): 97-112.
8. Marsh D, Zori R. 2002. Genetic Insights into Familial Cancers — Update and Recent Discoveries. Cancer Lett 181(2): 125-64.
9. Goujon P. 2001. From Biotechnology to Genomes: The Meaning of the Double Helix. NJ: World Scientific. 728p.
(Art by Fan Sozzi)