PAINLESS GENE EXPRESSION PROFILING: SAGE (SERIAL ANALYSIS OF GENE EXPRESSION)
(August 2004)
With the advent of the human genome project, a vast amount of information about genes and gene structure is suddenly at our fingertips. But this information is limited. Every cell within an organism has the same genetic composition (with the exception of its gametes), and yet, obviously skin tissue is very different from nervous tissue. The DNA sequence cannot provide information about these differences, which represent the next level of complexity and organization within an organism: DNA expression. Cells within a multicellular organism, such as ourselves, specialize to perform specific functions to increase the efficiency of the organism. Nerve cells, or neurons, express neuron-specific proteins that allow it to perform neuron duties. Skin, or epithelial cells, have their own specific proteins that enable their protective functioning. Both neuron and epithelial cell have the genes encoding for neural- and epithelial-specific proteins, but each cell only expresses the genes that it requires, and not other tissue-specific genes (Figure 1). In this way, a given DNA sequence only provides information about what could be, not what actually is.
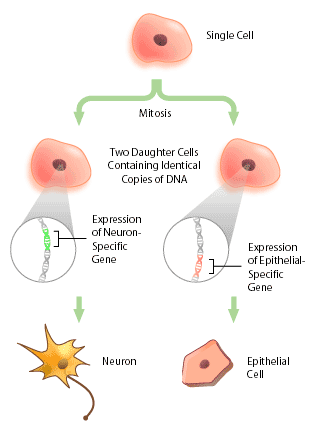
What Exactly is DNA Expression?
DNA expression refers to the study of how specific genes are transcribed at a given point in time in a given cell. A gene is transcribed into a messenger RNA (mRNA) transcript when the protein that is encoded by the gene is required by the cell. This occurs because DNA located in the nucleus, but all of the machinery necessary for translation, or producing proteins, resides in the cytoplasm. The cell resolves this problem by creating a copy of the gene (mRNA) that is capable of entering the cytoplasm through the nuclear pores. By examining which transcripts are present in a cell, it is possible to deduce which genes (and their related proteins) are expressed in a cell type, and at what time these are expressed.
In the past, DNA expression studies typically looked at only a few transcripts at any one time, due to the limitations of the techniques available [1]. But in recent years several new techniques have been developed that enable large scale studies of DNA expression; these can be used to create ‘expression profiles’. An expression profile is a characterization of the relative quantity of every transcript that is produced in any one cell type. One technique that has been used to generate expression profiles is SAGE (Serial Analysis of Gene Expression).
What is SAGE (Apart from a Spice)?
SAGE is a technique that allows rapid, detailed analysis of thousands of transcripts in a cell. The basic concept of SAGE rests on two principles: firstly, a small sequence of nucleotides from the transcript, called a ‘tag’, can effectively identify the original transcript from whence it came, and secondly, that linking these tags allows for rapid sequencing analysis of multiple transcripts. Imagine having thousands of transcripts to sequence – each sequencing event would take a certain amount of time to complete, and several thousand of these events would be necessary to identify each individual transcript. By linking the tags together, only one sequencing event is required to sequence every transcript within the cell, making the task of DNA expression profiling a much less daunting one [1,2].
Five Easy Steps and You Too Can Do SAGE
Figure 2 shows a schematic diagram of each of the steps in SAGE. First, a complimentary DNA strand, or cDNA, of each transcript in the cell must be generated. This is necessary, since mRNA is much less stable than DNA. The mRNA of eukaryotes is polyadenylated, meaning a poly(A) tail is added to the 3′ end of the final transcript. Therefore, a primer consisting of multiple ‘T’s can be made that will complimentary base pair with the poly(A) tail of every mRNAs in a cell. Once the primer has bound to the mRNA, the enzyme reverse transcriptase can make a DNA strand that is complimentary to the RNA. This DNA strand will then be converted to a double-stranded DNA molecule, which can then proceed to the next step.
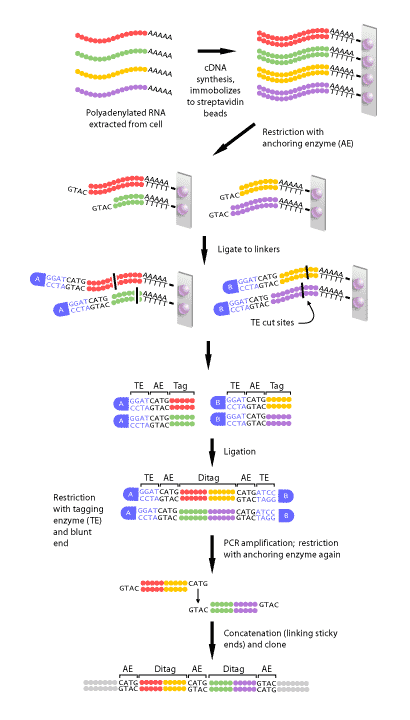
Once the cDNA has been created, it is then cleaved using an anchoring enzyme [1]. The anchoring enzyme is a restriction endonuclease that recognizes and cuts specific 4 base pair (bp) DNA sequences. Since this enzyme requires only 4 specific nucleotides, it cleaves DNA molecules often, resulting in every cDNA that has been generated being cut at least once. The cut cDNA is then bound to streptavidin beads by virtue of its multiple thymidine (‘T’s) at its 3′ end, thereby immobilizing it. At this point, we have multiple cDNAs bound to beads that are all of varying lengths, each with the same cut site and 5′ overhang, because the anchoring enzyme recognizes the same 4 bp in all transcripts, but these 4 bp occur at different points within each transcript.
The sample of bound cDNAs is then divided in half and ligated to either linker A or B. These linkers are designed to contain a type IIS restriction site. Type IIS restriction endonucleases cut at a defined distance up to 20 base pairs away from their recognition sites. The type IIS restrictive endonuclease, also called the tagging enzyme, cleaves the cDNA to release it from its bound bead. Blunt ends are then created, so that neither the 3′ nor 5′ end have overhanging single-stranded sequences. Once this is achieved, the cDNA tags bound to linker A and B are ligated to each other to create ditags. These ditags have linker A on one end, linker B on the other, and both transcript tags are adjacent to one another in the middle. These ditags are then amplified by PCR, using primers that are complimentary to sequence in either linker [1].
Once the ditags have been amplified, they are then cleaved using the anchoring enzyme again. This has two effects: first, it releases the linkers from either end of the ditag, leaving only sequence from the two tags. Secondly, it creates sticky ends, or 3′ and 5’ ends that have overhanging, single-stranded DNA that can complimentary base pair with single-stranded DNA of another ditag. In this way, all of the ditags generated are linked, or concatenated to produce one long string of tags. This collection of tags is then introduced into a vector to be cloned and sequenced[1].
The Problem with Gene Tags
There are several problems to be aware of when using SAGE. One that has proved to be a double-edged sword is the length of the gene tag. The tags generated during SAGE are extremely short (13 – 14 bp). If the tag derived is from an unknown gene, it is difficult to investigate its potential function using such a short sequence [3]. However, this hindrance could actually be advantageous, since isolating novel genes is often the ultimate goal for gene expression studies. Thus, SAGE could also be used as a “gene finding method”. In cancer research, for example, the most attractive feature of SAGE is its ability to evaluate the expression pattern of thousands of genes in a quantitative manner without prior sequence information. This has lead to analysis of differential gene expression of cancer cells and their normal counterparts, and identification of several novel genes that could potentially play a role in tumourigenesis [4].
Yet another problem with tags is tag specificity. There are instances in which multiple genes share the same tag, as there is an overlap in sequence between the two genes; this problem can be eliminated by using longer tag sequences. Increasing tag length to 18 bp in an attempt to increase tag specificity has been reported to yield a better representation of DNA expression [5].
Another downfall of the SAGE technique is that typeIIS restriction enzymes (typically BsmFI) do not always yield the same length of fragments. BsmFI should yield exact 14 bp tags, but, depending on the temperature, the length of fragments produced varies. Since two tags are ligated tail to tail, it is hard to ensure each tag is 14 bp long in a ditag of 28 bp – the ditag could be consist of a 12 bp and 16 bp tag, a 13 bp and 15 bp tag, or any variation thereof. This problem can be minimized by maintaining the temperature at 65° C [3].
One potential problem to be aware of is that certain species of mRNAs will not contain the enzyme recognition sequence. These transcripts cannot not be cleaved by the enzyme, and consequently, are not be included in the analysis. To avoid this problem, two different combinations of anchoring and tagging enzyme could be used and a gene expression profile created for each. The two profiles could then be correlated and compiled to represent the majority of the genes expressed within a cell accurately [3].
Conclusion
With the advent of DNA expression studies came the need for new technology. In the past, gene expression analysis had been restricted to examining only a few given genes at one time — SAGE, and other tests of its kind have eliminated these limitations. By providing a rapid means of determining all of the transcripts present within a cell, one that evades the trials and tribulations of large sample numbers, SAGE is capable of providing scientists with a functional profile of gene expression. These profiles can be used as a powerful tool in investigating a variety of transcriptional phenomena. Even now, these expression profiles are being used in diagnosing cancer susceptibility and identifying key genes related to disease [4].
References
1. Velculescu, V.E., Zhang, L., Vogelstein, B. and Kinzler, K.W. (1995). Serial analysis of gene expression. Science 270:484-7
2. Carulli, J.P., Artinger, M., Swain, P.M., Root, C.D., Chee, L., Tulig, C., Guerin, J., Osborne, M., Stein, G., Lian, J. and Lomedico, P. (1998). High throughput analysis of differential gene expression. Journal of Cellular Biochemistry Supplements 30/31:286-96
3. Yamamoto, M., Wakatsuki, T., Hada, A. and Ryo, A. (2001). Use of serial analysis of gene expression (SAGE) technology. Journal of Immunological Methods 250:45-66
4. Polyak, K. and Riggins, G.J. (2001). Gene discovery using the serial analysis of gene expression technique: implication for cancer research. Journal of Clinical Oncology 19:2948-58
5. Ryo, A., Kondoh, N., Wakatsuki, T., Hada, A., Yamamoto, N. and Yamamoto, M. (2000). A modified serial analysis of gene expression that generates longer sequence tags by nonpalindromic cohesive linker ligation. Analytical Biochemistry 277:160-2
(Art by Jiang Long – note that high res versions of image files available here)