PRESCRIPTION ANTIBIOTICS – HOW EXACTLY DO THESE DRUGS WORK
(August 2003)
Antibiotics are compounds that act to kill or inhibit the growth of bacteria1. The etymology of the term can be broken down into two roots: the prefix “anti-” meaning “opposed to” or “preventing” and “biotic” coming from the Greek word for life. In nature, various microbes and fungi secrete these compounds to gain an advantage in their microenvironment and it is from these very organisms that antibiotics are commonly use isolated [1].
The Discovery of Antibiotics
The stories of the discovery of antibiotics are dramatic and full of human interest, both on a global and personal scale. Two brief recounts are given below (please see reference 2 for an excellent overview of antibiotics).
Alexander Fleming is popularly thought to have been the discoverer of penicillin. He is certainly the first researcher to have recognized its potential. In 1928 Fleming discovered that a blue mold (Penicillium) was able to lyse bacterial Staphylococci cells. Fleming determined that Penicillium produced some compound that caused the bacterial cells to lyse. He called this compound penicillin. And how he came to these conclusions is a now famous story of fortuitous chance [3]. Fleming had returned to the lab after a holiday to discover that some culture plates of Staphylococci had become contaminated. It was an accidental growth of Penicillium, but luckily one he did not throw away. His further observation that the contaminating mold was able to kill Staphylococci led to his being awarded the Nobel Prize for Medicine in 1945. Howard Florey and Ernest Chain were also honoured with the Nobel Prize for developing a way to produce large quantities of penicillin.
Gerhard Domagk discovered the first sulfa drugs in 1932. The pharmaceutical company Bayer had hired him to work on the problem of infectious diseases caused by the bacterium Streptococcus pyogenes. In various trials designed to determine the effectiveness of various compounds for bacterial killing, Domagk discovered that a dye called prontosil rubrum prevented S. pyogenes infection in mice. In 1935 Domagk’s daughter was gravely ill due to S. pyogenes infection. The infection was advancing so aggressively that doctors were considering amputating her arm, but instead she was treated with prontosil rubrum (well before complete clinical trials of the drug had been completed). She made a full recovery. Domagk received the Nobel Prize in 1939.
Mechanism of Action
Being such an important medical compound the mechanisms by which antibiotics kill bacteria have been under scrutiny for decades, with such studies being instrumental in the design of new and improved compounds. There are three general modes of antibiotic activity: (1) interference with the cell wall, (2) interference with nucleic acid synthesis, and (3) interference with protein synthesis.
Interference with the Cell Wall
There is a multitude of ways to classify bacteria, but one of the more common methods is as either Gram-positive or Gram-negative cells [4]. These Gram designations are based on a differential staining assay, with the bacteria that stain dark being referred to as Gram-positive, while those that do not stain dark being referred to as Gram-negative. This difference in stain intensity and thus designation does have a physical basis, which is linked to the cell membrane. In every bacterial cell the plasma membrane encases the contents of the cell (referred to as the cytoplasm) and directly outside the plasma membrane is an additional exterior cell wall (See Figure 1).
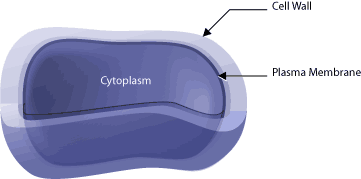
The plasma membrane of the cell is pressed tightly against the cell wall due to turgor pressure. It is the cell wall that determines the shape of the cells, the strength of which is provided by peptidoglycan [5] (the major structural component of the cell wall). Gram-positive bacteria have thick cell walls composed primarily of this substance; where as Gram-negative bacteria have multi-layer cell walls that are thinner than those of the Gram-positive cells. Peptidoglycan is a lattice-like macromolecule composed of repeated sugar units that are cross-linked together, with the “glycans” of peptidoglycan forming polysaccharide strands that run parallel to each other (See Figure 2). These polysaccharide strands are composed of alternating units of two sugars: N-acetylmuramic acid (commonly referred to as NAM) and N-acetylglucosamine (commonly referred to as NAG). Additionally, peptidoglycan contains two peptides that cross-link these long polysaccharide strands and are composed of alanine, glutamic acid, lysine and alanine. Tetra-peptides linking the polysaccharide strands are themselves linked by a penta-peptide of five glycine residues that runs from the lysine residue of one tetrapeptide to the terminal alanine residue of another tetra-peptide. Figure 2 shows how these three different components (the polysaccharide strands, the tetra-peptide and the penta-peptide) come together to form peptidoglycan and give this macromoleclue its strength.
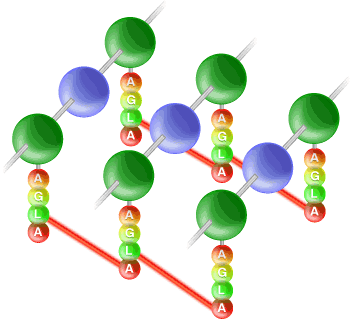
Peptidoglycan is synthesized in three stages in three different parts of the cell. Firstly, the NAM sugar is linked to the alanine, glutamic acid, lysine and alanine precursor in the cytoplasm, forming the basic subunit of peptidoglycan. Secondly, the NAM/peptide is linked to the NAG sugar at the cell membrane. And lastly, now at the cell wall, the newly synthesized peptidoglycan subunit is transferred to the growing point of the cell wall’s peptidoglycan by a bond between the old peptidoglycan and the new NAM-NAG disaccharide. Here the lateral cross-linking by the pentaglycine peptide can occur.
Antibiotics are active at every step of peptidoglycan synthesis. Among the most famous of the antibiotics to interfere with peptidoglycan synthesis are the b-lactams, of which penicillin is an example [6]. Penicillins resemble the terminal amino acids of the NAM+tetrapeptide precursor synthesized in the cytoplasm and actually bind the enzyme that catalyzes the pentaglycine cross-linking reaction. This binding prevents the cross-linking reaction from occurring and thus weakens the cell wall. Eventually, the turgor pressure of the cell causes the cell to lose its shape and eventually burst if the surrounding solution is hypotonic. Go here to see a short movie showing the activity of penicillin on Escherichia coli. On the flip side of this coin, bacteria can become resistant to penicillin by three strategies: the hydrolysis of penicillin, the acquisition of proteins with a reduced affinity for penicillin or the reduced uptake of penicillin. The hydrolysis of penicillin is catalyzed by enzymes called b-lactamases, which ring structure of penicillin and prevent it from mimicing the structure of the peptidoglycan precursor.
Interference with Nucleic Acid Synthesis
Antibiotics are frequently active against nucleic acid synthesis within the bacterial cell [1]. This includes inhibition or interference of deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) synthesis. The antibiotics that act in this manner are frequently analogs of essential metabolites of the cell, compounds that block DNA template activity during synthesis of new DNA, or compounds that block the transcription of DNA into RNA.
An example is the sulfonamides, commonly known as sulfa drugs, which are derivatives of dyes and resemble the compound folate. Folate is a coenzyme (a substance required for the proper functioning of enzymes) that is essential for cell growth and in bacterial cells is a precursor for the synthesis of amino acids and nucleic acids. Most bacteria synthesize folate from scratch, whereas mammalian cells cannot do this and must transport folate, which has been made by other sources, into their cells. This metabolic difference between bacterial cells and mammalian cells makes folate biosynthesis a convenient target for antibiotics, as this pathway is specific to bacterial cells. The sulfa drugs mimic one of the folate precursors, competing with the precursor for the enzyme involved in the next step of folate synthesis. If the normal folate precursor cannot bind the enzyme (because the sulfa drug is there), folate synthesis is blocked. Thus, the bacteria cannot synthesize the nucleic acids and some of the amino acids necessary for cell survival and perish.
The method by which an antibiotic affects nucleic acid synthesis is actually quite diverse. For example, the coiling of the bacterial chromosome can be attacked. It is quite a long structure and its great size requires that this it is efficiently packed into the cell by supercoiling. The tightly coiled DNA must be uncoiled and relaxed in order for the DNA or RNA polymerases to gain access to the DNA template, a process accomplished by enzymes known as topoisomerases. All three types of enzymes, the RNA and DNA polymerases and topoisomerases, are antibiotic targets. Drugs called quinolones target a topoisomerase known as ‘DNA gyrase’, which normally uncoils DNA by cutting the two strands and then passing a section of the double helix through the gap. Quinolones bind to the cut strands of DNA, preventing the re-annealing (gluing back together) of the cut DNA strands with the parent strands. An alternative is the nitroimidazoles, drugs that cleave the double-stranded DNA template by producing radical ions. Cleavage of the DNA template interferes with both DNA replication and RNA synthesis. Classes of drugs known as rifamycins actually specifically inhibit RNA synthesis by binding to the RNA polymerase and preventing the synthesis of the first dinucleotide of RNA. The binding of the RNA polymerase to the DNA template is not affected.
The example of sulfa drugs raises the important point that not all antibiotics are specific enough to be used in patients. Take the compound Actinomycin D, which targets DNA during RNA synthesis. It binds to DNA at guanine (G) and cytosine (C) basepairs and selectively inhibits RNA synthesis. Unfortunately, it is not selective for bacterial DNA and so is not used for treatment of bacterial infections. However, these compounds are very useful in the laboratory setting.
Interference with Protein Synthesis
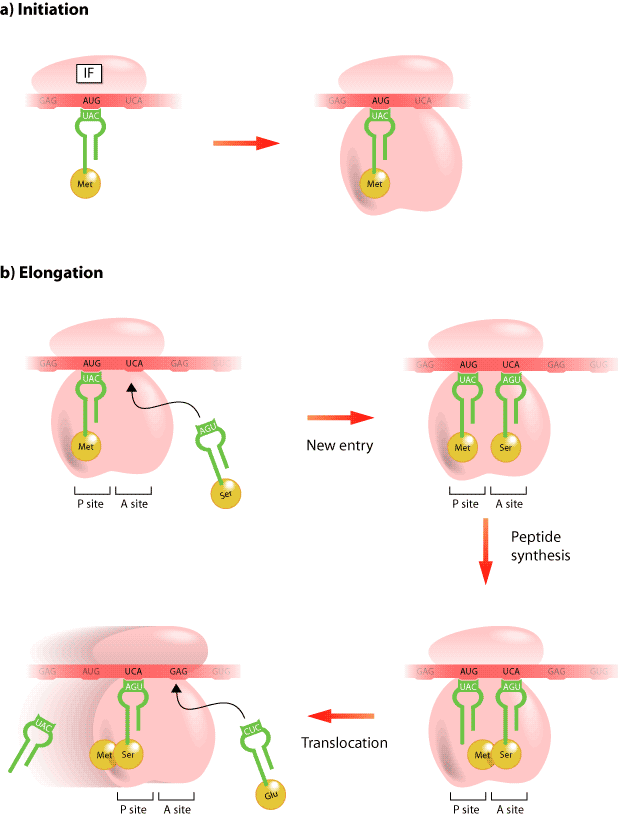
After DNA has been transcribed into messenger RNA (mRNA), this message is translated into protein (Figure 3). The process requires a ribosome, the mRNA, and a second type of RNA called transfer RNA (tRNA). It is the ribosome that is the cellular machine responsible for making the protein and it does this through two amino acid sites known as the “A” and “P” sites. For example, the first amino acid of the protein is carried to the “P” site by a tRNA that corresponds to a three-nucleotide sequence, known as a codon, within the mRNA sequence, while the codon sequence in the “A” site determines the identity of the next amino acid to be incorporated into the growing protein. As amino acids are brought to the ribosome by various tRNAs they are attached together and eventually an entire protein is created. There are antibiotics that inhibit the translational activity of the ribosome at various steps of protein synthesis [1]. For example, puromycin is a non-selective inhibitor of protein synthesis that is a mimic tRNA. It is incorporated into the ribosome at the “A” site and accepts the growing polypeptide chain by formation of a peptide bond, but it blocks the addition any more amino acids. Alternatively, streptomycin causes the incorporation of incorrect amino acids at the “A” site of the growing polypeptide, whereas, tetracyclines completely block protein translation by binding to a ribosomal subunit.
The Evolution of Antibiotic Resistance
There are resistance mechanisms for each of the antibiotics described above. Most often resistance results from either a change in a protein structure of the bacterium, an inactivation of the antibiotic drug, the prevention of antibiotic accumulation, or the block of its entry into a cell [7]. The increased development of “anti-antibiotic” strategies in bacterial cells is actually a result of the use of antibiotics. This is because the use of antibiotics creates a strong selective pressure that favors those bacteria that acquire such mechanisms of resistance. In a population of bacteria that are sensitive to an antibiotic, its use will prevent those bacteria from leaving descendant or daughter cells. However, in any population of bacteria there will be occasional random mutations in the protein sequence of the various enzymes within any particular cell. If one of these mutations gives rise to a protein that is impervious to the action of the antibiotic, that cell will survive and produce descendant cells that are also resistant to the activity of the antibiotic. In fact, the biology of bacteria provides ideal opportunities for these chance occurrences of resistance. Since, under ideal conditions an E. coli bacterium can divide every two hours, the chance it making a beneficial mistake is high enough for such resistances to occur and flourish due to the antibiotic selection. Random mutation is not the only way that a bacterial cell can acquire resistance to antibiotics. Bacteria can also take up foreign DNA from their environment and from other bacteria. Thus, if a bacterium of one species is resistant to an antibiotic it is possible that the DNA encoding the resistant protein may be transferred to bacteria of another (formerly sensitive) species.
The acquisition of resistance in bacteria is a serious problem [1,7]. Some antibiotics are no longer useful for treating infections because bacterial resistance to the antibiotic has spread worldwide. Bacterial resistance to antibiotics can develop and spread very quickly, rendering an antibiotic ineffective within only a few years. The development of new antibiotics is both expensive and time-consuming and in some cases it appears that bacteria are developing resistance to antibiotics faster than scientists can develop them. It is hoped that our continued drive to understand how antibiotics work will keep these useful drugs available.
Additional Reading
1. McDermott PF, et al. 2003. Antimicrobials: Modes of Action and Mechanisms of Resistance. Int J Toxicol 22(2): 135-43.
2. Amyes S. 2001. Magic Bullets, Lost Horizons: The Rise and Fall of Antibiotics. London/New York: Taylor & Francis. 262 p.
3. Chopra I, et al. 2002. Exploiting current understanding of antibiotic action for discovery of new drugs. J Appl Microbiol 92 Suppl: 4S-15S.
4. Scott GM, Kyi MS. 2001. Handbook of Essential Antibiotics. Amsterdam: Harwood Academic. 117p.
References
1. Walsh C. 2003. Antibiotics: Actions, Origins, Resistance. Washington, DC: ASM Press. 335p.
2. Birch B. 1990. Alexander Fleming: The Bacteriologist who Discovered Penicillin, A Miracle Drug that has Saved Millions of Lives. Toronto: Irwin Pub. 64p.
3. A History of Antibiotics [videorecording]: A Presentation of Films for the Humanities & Sciences. Princeton, NJ: Films for the Humanities and Sciences, c2000. 1 videocassette (45 min.).
4. Beveridge TJ. 2001. Use of the gram stain in microbiology. Biotech Histochem 76(3): 111-8.
5. Shockman GD, Barrett JF. 1983. Structure, function, and assembly of cell walls of gram-positive bacteria. Annu Rev Microbiol 37: 501-27.
6. Ghuysen JM, et al. 1996. Penicillin and beyond: evolution, protein fold, multimodular polypeptides, and multiprotein complexes. Microb Drug Resist 2(2): 163-75.
7. Walsh C. 2000. Molecular mechanisms that confer antibacterial drug resistance. Nature 406(6797): 775-81 .
(Art by Jiang Long – note that high res versions of these image files are available here)