PRIONS: INFECTIOUS PROTEINS REPSONSIBLE FOR MAD COW DISEASE
(August 2003)
Prions and prion diseases have been widely discussed in the media in recent years. The interest in prions and prion diseases was stimulated by the outbreak of bovine spongiform encephalopathy (BSE or Mad Cow Disease) in Europe in the mid-nineteen-nineties. This is a relatively new area of study. What exactly has been discovered about prions and prion diseases?
What Are Prions?
Prions are proteins found on the plasma membrane (the membrane that surrounds a cell and defines its physical boundary). In mammals, prions are found in the highest concentration in cells of the central nervous system. In mammals and yeast, there are several genes coding for different prions. The function of normal prions (denoted PrPC) is unknown. Aberrant or mutant prions (denoted PrPSc) are thought to be the causative agents of a set of neurological disorders, among which are BSE in cows and Creuzfeldt-Jacob Disease (CJD) in humans. The term prion comes from “proteinaceous infectious particles”. The term was coined during early research of the sheep disease called scrapie. During that time, all that was known about the particle that caused scrapie was that it was a protein and that it was infectious.
Prions have the same system of organization as other proteins. They may have up to four levels of organization. The primary structure of a protein is the amino acid sequence that makes up the protein. The secondary structure of a protein is the local organization of the primary sequence of the protein -helical or -sheet formations. The tertiary structure of a protein is the overall shape of the protein, caused by interactions between the various local structures. Some proteins have a quaternary level of organization, which is defined by the interactions between the tertiary structures of two or more protein subunits.
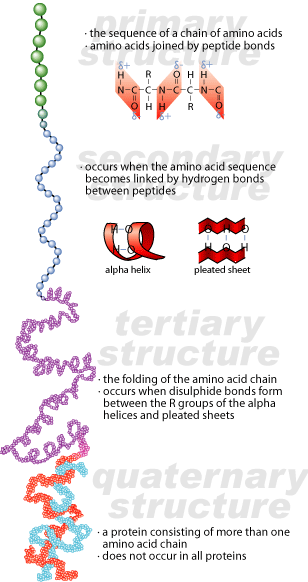
How Can Proteins Be Infectious?
Prions have one characteristic that makes them unique: they can exist in two different conformations at the level of secondary structure. There is an alpha-helical portion of secondary structure in a normal prion that is refolded into beta-sheet formation in an aberrant prion. The primary structure of the aberrant prion remains the same, but its secondary structure is different. As a result, its tertiary structure will be different as well.
Infection of normal cells may occur when an aberrant prion acts as a template for the refolding of a normal prion into a new aberrant prion. It is thought that there is at least one more protein involved: the as-yet-unidentified Protein X. This protein is believed to mediate the folding from a normal into an abnormal prion. When proteins are synthesized inside of a cell there are other special proteins (known as chaperones) that help in this process. Chaperones are proteins that bind to the newly synthesized protein or protein subunit, in order to ensure that the protein is properly folded into its secondary or tertiary structure. It has been hypothesized that Protein X is a type of chaperone.
The concept that a protein can transmit information (i.e. protein structure) is novel. At the time that this idea was proposed, it was truly radical; it conflicted with many years of evidence showing that only DNA was capable of transmitting such information.
This does not mean that prion diseases cannot be genetic. If an individual happens to carry a gene that codes for a mutant prion, then all of the prions that their cells produce will be the abnormal, disease-causing prions. One such disease is familial CJD.
The CJD-BSE Connection
The prion disease of humans that causes the most concern is variant CJD (vCJD). The cause of this disease has not yet been determined. One theory is that humans may become infected with aberrant prions after ingesting meat from cows infected with BSE. This would require that aberrant prions from one species (cows) could serve as a template for refolding normal prions of another species (humans). This jump from one species to another involves crossing what is referred to as the “species barrier”. In a laboratory setting, prions have been shown to cross the species barrier. The onset of disease in these cases, however, is delayed compared to cases in which the infecting (aberrant) prions are from the same species as the new host.
![prions[1]-GIF.gif](https://www.scq.ubc.ca/wp-content/uploads/2006/08/prions[1]-GIF.gif)
It is believed that the origin of the BSE outbreak in Europe (and especially Britain) was due to aberrant sheep prions crossing the species barrier to infect cows. During the 1970’s there were changes made to the process used to render offal for a livestock feed additive. These changes created conditions that allowed prions to survive the rendering process. The sheep disease known as scrapie is a prion disease. It is believed that prions from scrapie-infected sheep survived the rendering process and were passed on to cows that ingested infected feed. Once the prions were established in the livestock population the feed could have acted as a reservoir of infective prions. The practice of using rendered offal as an additive to livestock feed has been discontinued. Whether or not bovine prions can be passed to humans is unclear. Human prion diseases like vCJD have long incubation times (typically more than 12 years). The details of the transmission of prions from one species to another have not been fully elucidated.
How do Aberrant Prions Cause Disease?
The mechanism by which aberrant prions cause disease pathology has not been established. Several studies have been performed that may shed some light on the area. The fact that there are so many prions found in the cells of the Central Nervous System is thought to imply that these proteins are of some importance. This theory has been countered by an experiment using knockout mice. Mice were created with both prion alleles disturbed, so that the mice had no prions (PrPo/o) in their cell membranes. These mice did not show any sign of illness. When a total lack of protein does not cause disease, it is inferred that the protein is unnecessary for maintaining the health of the organism? This assumption has been criticized in this case because results from experiments on mice cannot necessarily be extrapolated to humans. It is also possible that the tests used to determine the presence of disease in mice are not sensitive enough to pick up any changes caused by the absence of prions.
The knock-out mice have been used to support the theory that aberrant prions are used as templates for the refolding of normal prions. The prion knock-out mice cannot be infected by mutant prions. Healthy cells can be exposed to mutant prions without any ill effects. This finding supports the theory that infection is spread by the refolding of normal proteins.
Unsolved Mystery
More studies must be done before it can be determined whether disease is caused by the lack of normal prions, or by the presence of aberrant prions. This is only one of the questions researchers are currently trying to answer. There is currently much research being conducted on the mechanism of the transformation of normal prions into mutant prions. The goal of these studies is to design drugs that can interfere with this refolding process. Information about the prion neurodegenerative diseases could be useful in research into other non-prion-related neurodegenerative diseases (such as Alzheimer’s disease).
References
1. University of Leicester, Microbiology & Immunology Department. Prion Diseases
2. University of Illinois at Urbana-Champaign, Department of Animal Sciences. BSE Information at UIUC
(Art by Jen Philpot)