METATRANSCRIPTOME OF GUT MICROBIOTA IN WOOKIEES WITH SEVERE INFLAMMATORY BOWEL DISEASE REVEALS DISTINCT PATHOGENIC COMMUNITY COMPOSITION AND FUNCTIONAL PROFILE
Just another reminder that this paper will be published in print format in the fall, along with selected SCQ pieces from the last 7 or so years. If you want to submit your creative science pieces with a mind to make the first issue of this new journal, then please visit here for more information.

Annals of Praetachoral Mechanics. (2014). Vol 1. pp49-58 pdf download
ABSTRACT
As the Galactic Empire continues to invade Kashyyyk, surviving Wookiees are finding refuge with humans on Alderaan. However, displaced Wookiees are exhibiting severe inflammatory bowel disease (IBD) -like symptoms. We hypothesized that dysbiosis in the microbiota of the gut exists between IBD-afflicted refugee Wookiees compared to healthy Wookiees that remain on Kashyyyk. To test this, we conducted a metatranscriptomic survey of the fecal microbiome in the two Wookiee populations. Our results show decreased microbial diversity in IBD fecal samples. Also, IBD patients showed a significantly increased population of Proteobacteria and reduced population of Lachnospiraceae and Firmicutes. We also found that Fusobacteriaceae and Clostridiaceae were unique to the microbiome in IBD patients. Functional analysis of the microbiome revealed significant enrichment in secondary metabolite biosynthesis and defense mechanism pathways. Additionally, considerably more transcripts were assigned with an unknown function in IBD samples. Taken together, we propose that the changes in community structure and function observed in IBD patients are indicative of a severely dysfunctional gut microbiome. Considering the drastic change in environment from Kashyyyk to Alderaan, including food variety, it is possible that this is a causal factor in IBD-afflicted Wookiees. Further investigation is needed to reveal the underlying mechanism of this microbiome discrepancy and possible treatment.
Key words: Transcriptomics, inflammatory bowel disease, Wookiee, microbial community structure, microbial community function
Introduction
The ongoing invasion by the Galactic Empire on the jungle planet Kashyyyk, native home to the humanoid species Wookiees, has led to a diaspora of Wookiees seeking refuge on planet Alderaan. Currently, the Alderaanian planetary government has provided a home to over 2000 Wookiees, mainly females and children who have survived battle. However, many of the refugee Wookiees have developed a severe gastrointestinal disorder. The symptoms of this disorder are very similar to inflammatory bowel disease (IBD) in humans. Common symptoms in Wookiees include severe abdominal pain, diarrhea, excessive hair loss, bloody vomit and stool, loss of vision, severe weight loss, and eventually death. Considering the severity of this illness, the Royal Alderaanian Institute of Genomics is collaborating with Wookiee researchers at the Kashyyyk Health Congress in Rwookrrorro, who remain in hiding on planet Kashyyyk. This study represents a collaborative effort to determine a causal factor for this illness and provide research direction for treatment
In humans, IBD is an acute or chronic condition characterized by gross intestinal inflammation which affects more than 3.6 million humans on Alderaan [1, 4]. The two main forms of IBD are Crohn’s disease and ulcerative colitis [2]. IBD has been associated with environmental, genetic and dietary factors and there is increasing evidence that the microbial community in the gut has a direct relationship with inflammation in the GI tract [2, 3, 4].
The human gut is home to 1014 microorganisms [8]. Microbial diversity exists between individuals in a population depending on genotype, age, health status and diet [5]. Human research has shown that these bacteria are beneficial for nutrient metabolism, immune function and repressing the growth of harmful microorganisms [4, 5]. In humans, studies show that IBD is associated with decreased gut microbial diversity, a decreased population in bacteria phylum Firmicutes and increased Proteobacteria populations [4, 9]. To elucidate the underlying cause of this IBD-like disorder in Wookiees, we suggest that a difference exists between the population of gut microbiota between IBD-affected Alderaan-residing Wookiees (henceforth IBD-afflicted) and healthy control Kashyyyk-residing Wookiees. We also suspect that there will be dysbiosis of the gut community between these two groups. To test for this, we will be sampling the gut microbiota by taking fecal samples from affected Alderaan-dwelling Wookiees and their unaffected counterparts living in Kashyyyk. In order to gain insight into the gut microbial diversity, we extracted total RNA and pyrosequenced transcripts to evaluate the active microbial community, and we purified and sequenced prokaryotic mRNA to map the functional abilities of the gut microbiome in both IBD and healthy Wookiees. To our knowledge, this is the first study comparing the microbiome in two Wookiee populations.
Methods
Sample collection
This study is a collaborative project between the Royal Alderaanian Institute of Genomics and the Kashyyyk Health Congress. Alderaan and Kashyyyk teams were responsible for identifying 10 Wookiee individuals for study on each planet. After informed consent was provided, fecal samples were collected from 10 IBD afflicted individuals at the Juranno General Hospital Intensive Care Unit on Alderaan and 10 healthy volunteers from the last remaining Wookiee stronghold in Rwookrrorro on Kashyyyk. Healthy individuals were screened for intestinal disorders or recent antibiotic treatment. Sterile containers with 30 mL of saline buffer were used for collection and samples were stored at -80°C until required. Previous human studies have determined that microbiota along the intestinal tract are correlated with the microbiota of fecal matter, eliminating the need for invasive ileum sampling [5].
Total RNA extraction and mRNA purification
Total RNA was extracted from 1 g of frozen sample using the RiboPure-Bacteria kit (Ambion) following the manufacturer’s instructions. Each sample was separated into rRNA for phylogenetic analysis and into mRNA for functional analysis. Full methods for separation are found in Turnbaugh et al. 2010 [6]. Briefly, mRNA was amplified using the MessageAmp II-Bacteria kit (Ambion), where bacterial mRNAs were adenylated with E.coli poly(A) polymerase and linearly amplified. Both total RNA and purified mRNA were converted to cDNA with SuperScript II reverse transcriptase (Invitrogen).
Pyrosequencing
cDNAs (5 ug cDNA samples) were sequenced at Princess Leia Life Sciences Centre (Aldera) on a Roche GS FLX sequencer. Sample transport from Kashyyyk was arranged via delivery on the Millennium Falcon light freighter through a generous donation of time and expertise by H. Solo Enterprises.
Community profiling
Profiling of sequence data was performed to identify 16SrRNA tags (taxonomy) and mRNA tags (function), as described in Urich et al. 2008 [7]. Taxonomic tags were compared with 5000 pre-selected rRNA sequences from the Kashyyykian Centre for Biotechnology Information (KCBI) environmental metagenome database. Species identification were made using BLAST with a 100 bp fragment and a 97% sequence homology cut off. Large Subunit rRNA and 23S rRNA were not included in the analysis. Taxonomic relationships were inferred from 16S rRNA sequences using the recently developed Kashyyykian Prokaryotic Tree of Life tool (KProTol) [8]. Taxonomy was provided for the genus level where possible. All unassigned rRNA tags were labelled as such. The Shannon Index of Biodiversity was also computed for IBD and healthy fecal samples.
Functional profiling
cDNA sequences reverse transcribed from the enriched mRNA pool were aligned to the KCBI protein database and phylogenetically binned using MEGAN. As in Urich et al. 2008 [7], the MG-RAST (Meta Genome Rapid Annotation using Subsystem Technology) and the SEED database were used to predict unclassified protein hits into broad metabolic pathways (e.g. nitrogen metabolism, carbohydrate metabolism etc). Transcripts binned into each metabolic pathway group were compared between healthy and diseased fecal samples such that the relative abundance of each pathway could be compared between samples.
Results
Sequencing
Pyrosequencing of the 10 healthy fecal samples produced an average of 701,457 reads between 75 and 700 bp in size at an average read length of 264 bp. Pyrosequencing of the 10 IBD afflicted fecal samples produced an average 754,132 reads between 80 and 732 bp and an average read length of 254 bp. Of the cDNAs sequenced, 18.9% and 17.9% corresponded to 16S cDNA representing the active bacteria in the healthy and IBD datasets, respectively, All cDNAs that did not result in a hit in the KCBI rRNA database (132,575 and 134,989 reads from healthy and IBD samples, respectively) were compared to the KCBI protein database where about 48% of these reads found known homologues.
Taxonomic assignment of 16S cDNA
The KProTol tool produced a microbial community structure for pooled healthy vs. IBD fecal samples (Figure 1). The two largest phyla represented in each dataset of healthy and IBD-afflicted Wookiees were Firmicutes and Bacteriodetes. In healthy individuals, this corresponded to 49% and 31%, respectively, of the 16S rRNA transcripts. Also, Proteobacteria made up the next largest represented phylum (4.9%), with Actinobacteria in relatively small abundance (0.6%) and unassigned reads corresponding to 14.5% of the healthy individuals dataset. At the phyla level, the IBD dataset showed a significantly reduced (p<0.05) Firmicutes abundance (41%), while the abundance of the Bacteroidetes was comparable (29%) to the healthy dataset. The Proteobacteria were significantly (p<0.05) enriched in the IBD dataset, due almost entirely to a 60-fold increase in the Proterobacteria family Enterobacteriaceae. At the family level, Lachnospiraceae (Firmicutes) were also significantly reduced (p<0.05) in the IBD samples (Fig 1). Two families, Fusobacteriaceae and Clostridiaceae were unique to the IBD dataset (Fig 1). The relative abundance of the Firmicutes family, Ruminococcaceae (7.9-10.1%), were similar among both datasets (Fig 1). Within the Bacteriodetes, the families Bacteroidaceae (8.1-9.8%), Prevotellaceae (4.2-6.4%) and Rickenellaceae (2.8-3.2%) had similar relative abundance in both IBD and healthy individuals (Fig 1). Microbial diversity, as measured by the Shannon index, was significantly lower (p<0.05) in the IBD samples (H=1.1) vs. healthy samples (H=2.3).
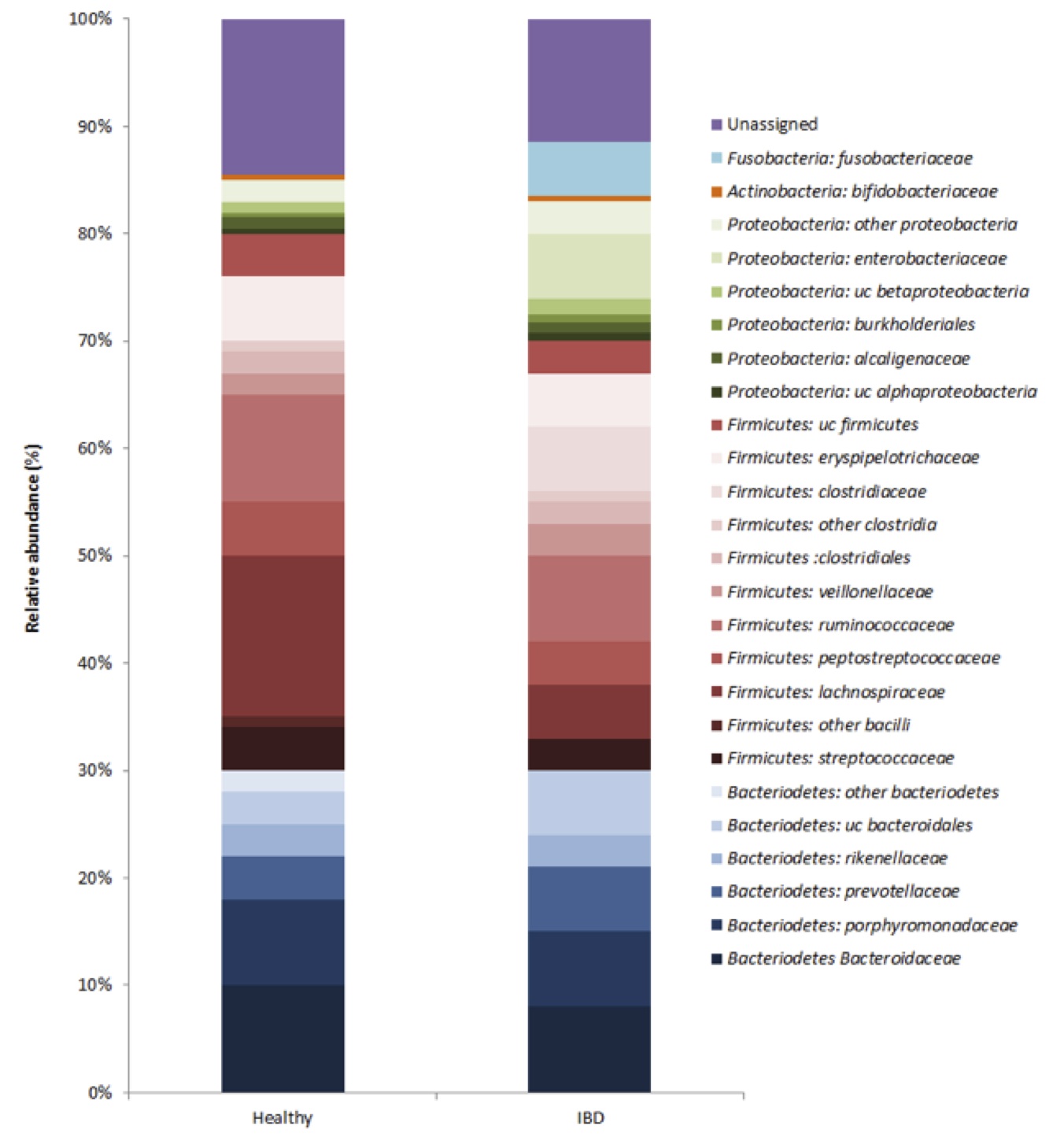
Figure 1 (Click to Enlarge): Composition of active microbiota in healthy and IBD Wookiee fecal samples
Functional assignment of mRNAs
Known and MG-RAST-predicted functional assignment of mRNAs in healthy and IBD afflicted Wookiees are shown in Figure 2. In general, functional pathways did not differ significantly between both data sets in most cases. Overall, energy production and conservation, carbohydrate transport and metabolism, and lipid transport and metabolism, were well represented in the datasets. However, significant (p<0.05) overrepresentation of secondary metabolite biosynthesis and defense mechanism pathways were found in IBD afflicted patients, with secondary metabolite biosynthesis and defense mechanisms being 4 and 5 times higher, respectively, in IBD vs. healthy samples. Additionally, IBD samples contained 2.5 times more transcripts of unknown function relative to healthy individuals.
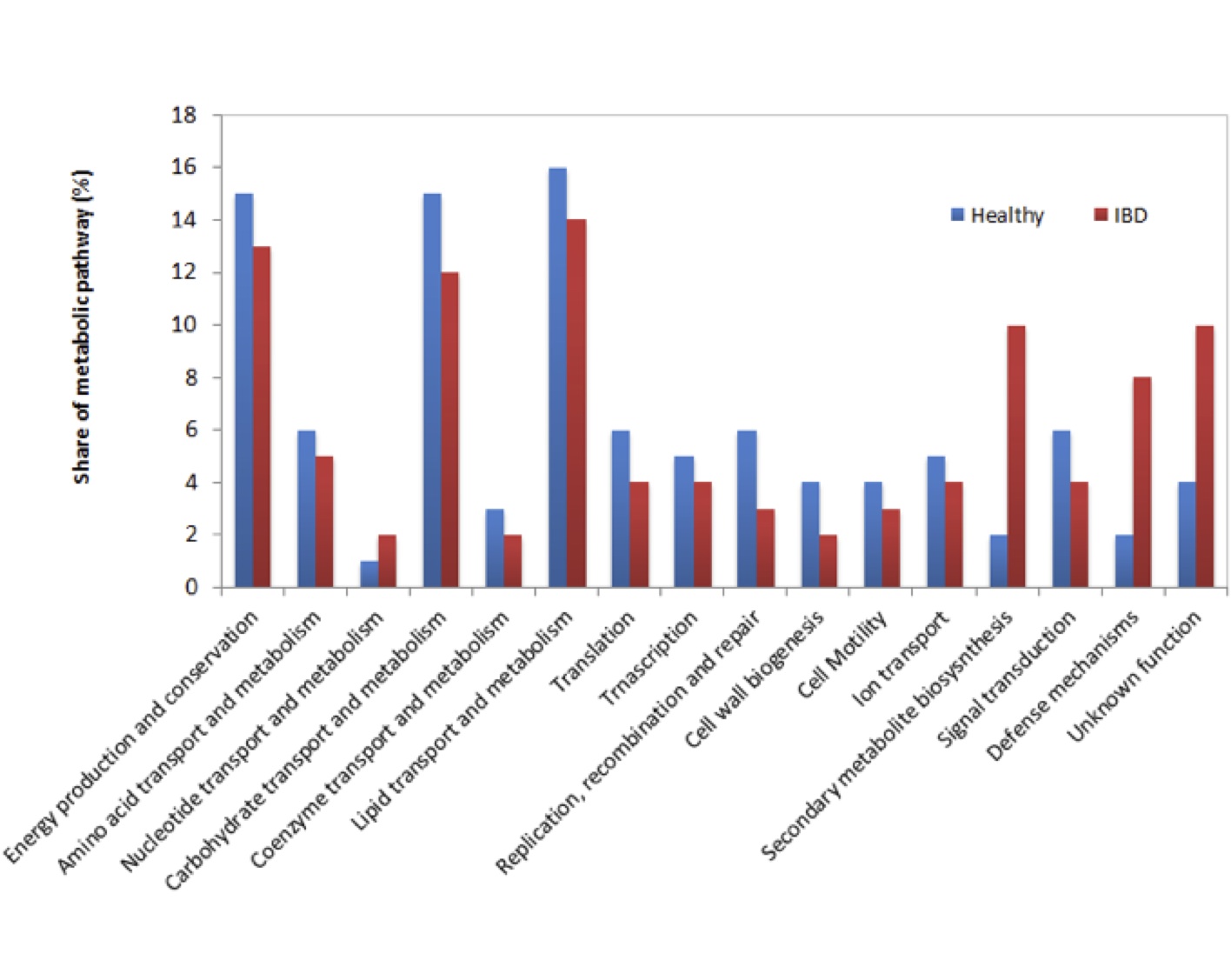
Figure 2 (Click t Enlarge): Functional analysis of mRNA tags from healthy and IBD Wookiee fecal samples samples assigned to functional categories. Asterisks represent significant differences (p<0.05)
Discussion
Current research in humans has revealed that gut microbial communities require a delicate balance to maintain health of the host [5, 9, 10, 11]. In IBD patients the host-microbe intestinal homeostasis may be in ‘dysbiosis,’ and in the case of IBD-afflicted Wookiee refugees on Alderaan, this dysbiosis may be proving fatal.
Our results point to several examples of dysbiosis in the gut of IBD-afflicted Wookiees. The decreased microbial diversity found in IBD fecal samples corresponds with studies done in humans [4, 12]. Additionally, the predominant phyla in both datasets are the Firmicutes and Bacteroidetes, similar to human gut microbiota [13], though IBD afflicted Wookiees showed a significantly reduced population of Firmicutes (Fig 1). Within the Bacteroidetes phylum, the relative abundance Bacteroidaceae, Prevotellaceae and Rickenellaceae were similar between datasets, with pectin/cellulose degradation function often ascribed to these families [10], suggesting these families may not be related to IBD pathogenesis. Decreased relative abundance of Lachnospiraceae, as seen in Wookiee IBD-afflicted fecal samples, has been reported in previous human studies [14], and may indicate that for IBD-afflicted Wookiees, this bacterial family has beneficial effects for host gastrointestinal health, but further study is required.
Complex interactions between the host immune system, host genotype and bacteria in the GI tract are believed to be responsible for the pathogenesis of IBD, although the underlying mechanisms remain unclear [11]. In this study, we present data showing three families either appearing in higher numbers in Wookiee IBD fecal samples or that are unique to the Wookiee fecal samples. Specifically, our analysis shows that Enterobacteriaceae appear to be enriched in IBD-afflicted Wookiees, while Fusobacteriacae and Clostridiaceae are unique to the Wookiee fecal samples (Fig 1). Enterobacteriaceae are gram-negative anaerobes that, through in vitro studies, have shown the ability to invade epithelial cells and induce granulomas [4]. Fusobacterium are also gram-negative anaerobes and are normally found in the oral cavity and occasionally the gut. Experimental models show that Fusobacterium varium can induce colonic mucosal erosion rectal enema [4]. The invasive nature of these bacteria may be a causal factor in IBD [4]. Segmented Filamentous Bacteria (Clostridiaceae, SFB) are an obscure and uncultivated bacterial species [5]. Mouse models show that SFB is involved in inducing colitis and significantly induce IgA response in humanoids [5]. Colonization of these bacteria in the GI tract is temporally related to the development of immune system in hosts [5]. These three pathogenic bacterial families present in IBD fecal samples may suggest involvement in the severe IBD-related symptoms.
The functional profile presented in Figure 2 demonstrates that for most of the functional groups identified, there were no major differences between IBD and healthy Wookiees. Most of the functional groups that were sequenced corresponded to energy production and conservation, carbohydrate transport and metabolism, and lipid transport and metabolism, suggesting that the functional role of the gut microbiota is largely based on energy production and nutrient processing, as has been found elsewhere [10]. The observed increase in secondary metabolite biosynthesis in IBD fecal samples provides evidence that the microbial community in IBD patients is over producing secondary metabolites, which can exhibit host toxicity if the metabolite is both toxic and produced in high volumes [16]. Additional sequencing is required to identify possible gene products from secondary metabolite biosynthesis that may play a role in Wookiee IBD pathogenesis. Defence mechanism metabolism was also highly elevated in IBD samples (Figure 2), which may be a microbial defense response to a host immunoresponse to IBD-related pathogenesis. Transcripts with no known function were also highly enriched in IBD fecal samples vs. healthy samples. This is the first result of its kind to be reported for IBD, although in this case there is an unusual severity in IBD-afflicted Wookiees transplanted to Alderaan. We can only speculate that these transcripts may be involved in the severity of the IBD in the Wookiees. Increased efforts to identify the gene products from these transcripts are currently underway.
It has been well documented that diet can directly affect the microbial community in the GI tract of humans [4]. Considering the drastic change from their planet Kashyyyk in diet and in the general environment, it is likely that IBD in Wookiees has been a result of these contributing factors. The IBD symptoms in Wookiees are therefore likely a result of a combination of the reduction in microbial diversity, increased Enterobactiaceae, Fusobacteriaceae and Clostridiaceae and a decreased Lachnospiraceae. This dysbiosis is supported by the relative increase of mRNA transcripts affiliated with secondary metabolite biosynthesis, defense mechanism pathways and a hitherto unknown functional pathway. The unknown functional pathways require further investigation as the underlying severe pathogenesis of the Wookiees may be directly related to this unique result. Human clinical trials have indicated that various probiotic treatments of live microbial supplements shows efficacy in IBD patients [5][6]. Since the intestinal bacteria are thought to comprise an important environmental factor in the pathogenesis of IBD, a more thorough understanding of the host-microbe intestinal dysbiosis is needed for elucidating the underlying mechanisms of IBD and for the treatment or prevention of this disease.
Conclusion
Overall, we suggest that the discrepancy between microbial populations may be causing the inflammatory bowel disease-like symptoms in Wookiees currently inhabiting Alderaan. The change in food variety and diet on Alderaan may be the culprit for afflicted individuals, although further investigation is required to confirm this. Future studies should investigate treatment options that have proven successful in human trials, such as probiotic supplements [5]. Future studies should determine the causal mechanism and transcripts involved in the pathogenesis of IBD in Wookiees.
Literature Cited
1. Şengül, N., Işık, S., Aslım, B., Uçar, G., & Demirbağ, A. E. (2011). The effect of exopolysaccharide-producing probiotic strains on gut oxidative damage in experimental colitis. Digestive diseases and sciences, 56(3), 707-714.
2.Baumgart, D. C., & Carding, S. R. (2007). Inflammatory bowel disease: cause and immunobiology. The Lancet, 369(9573), 1627-1640.
3. Centers for Disease Control and Prevention CDC. Inflammatory Bowel Disease. Division of Public Health. Accessed at www.cdc.gov/ibd/, 2014.
4. Kostic, A. D., Xavier, R. J., & Gevers, D. (2014). The Microbiome in Inflammatory Bowel Diseases: Current Status and the Future Ahead.Gastroenterology.
5. Peterson, D. A., Frank, D. N., Pace, N. R., & Gordon, J. I. (2008). Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell host & microbe, 3(6), 417-427.
6. Turnbaugh, P. J., Quince, C., Faith, J. J., McHardy, A. C., Yatsunenko, T., Niazi, F., … & Gordon, J. I. (2010). Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins.Proceedings of the National Academy of Sciences, 107(16), 7503-7508.
7. Urich, T., Lanzén, A., Qi, J., Huson, D. H., Schleper, C., & Schuster, S. C. (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One, 3(6), e2527.
8. Seymour, S, Anderson, D, et al. (2013) The Integrative Future of Taxonomy. Academic Press. 59(8), 25-28.
9.Berry, D., & Reinisch, W. (2013). Intestinal microbiota: A source of novel biomarkers in inflammatory bowel diseases?. Best Practice & Research Clinical Gastroenterology, 27(1), 47-58.
10. Gosalbes, M. J., Durbán, A., Pignatelli, M., Abellan, J. J., Jiménez-Hernández, N., Pérez-Cobas, A. E., … & Moya, A. (2011). Metatranscriptomic approach to analyze the functional human gut microbiota. PloS one, 6(3), e17447.
11.Reiff, C., & Kelly, D. (2010). Inflammatory bowel disease, gut bacteria and probiotic therapy. International Journal of Medical Microbiology, 300(1), 25-33.
12. Gevers, D., Kugathasan, S., Denson, L. A., Vázquez-Baeza, Y., Van Treuren, W., Ren, B., … & Xavier, R. J. (2014). The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell host & microbe, 15(3), 382-392.
13. Mahowald, M. A., Rey, F. E., Seedorf, H., Turnbaugh, P. J., Fulton, R. S., Wollam, A., … & Gordon, J. I. (2009). Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proceedings of the National Academy of Sciences, 106(14), 5859-5864.
14.Frank, D. N., Amand, A. L. S., Feldman, R. A., Boedeker, E. C., Harpaz, N., & Pace, N. R. (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences, 104(34), 13780-13785.
15. Medema, M. H., Blin, K., Cimermancic, P., de Jager, V., Zakrzewski, P., Fischbach, M. A., … & Breitling, R. (2011). antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic acids research, 39(suppl 2), W339-W346.
16. Chewbacca, R.H. 2010. Toxicity of secondary metabolites in humanoids. Annals of Praetachoral Mechanics 2(1), 48-61.
(Please note that this paper is not real)