INTRODUCTION TO PHYLOGENETICS.
From the time of Charles Darwin, it has been the dream of many biologists to reconstruct the evolutionary history of all organisms on Earth and express it in the form of a phylogenetic tree. Phylogeny uses evolutionary distance, or evolutionary relationship, as a way of classifying organisms (taxonomy).
Phylogenetic relationship between organisms is given by the degree and kind of evolutionary distance. To understand this concept better, let us define taxonomy. Taxonomy is the science of naming, classifying and describing organisms. Taxonomists arrange the different organisms in taxa (groups). These are then further grouped together depending on biological similarities. This grouping of taxa reflects the degree of biological similarity.
Systematics takes taxonomy one step further by elucidating new methods and theories that can be used to classify species. This classification is based on similarity traits and possible mechanisms of evolution. In the 1950s, William Hennig, a German biologist, proposed that systematics should reflect the known evolutionary history of lineages, an approach he called phylogenetic systematics. Therefore, phylogenetic systematics is the field that deals with identifying and understanding the evolutionary relationships among many different kinds of organisms
Phylogenic relationships have been traditionally studied based on morphological data. Scientists used to examine different traits or characteristics and tried to establish the degree of relatedness between organisms. Then scientists realized that not all shared characteristics are useful in studying relationships between organisms. This discovery led to a study of systematics called cladistics. Cladistics is the study of phylogenetic relationships based on shared, derived characteristics. There are two types of characteristics, primitive traits and derived traits, which are described below.
Primitive traits are characteristics of organisms that were present in the ancestor of the group that is under study. They do not indicate anything about the relationships of species within a group because they are inherited from the ancestor to all of the members of the group. Derived traits are characteristics of organisms that have evolved within the group under study. These characteristics were not present in the ancestor. They are useful because they can help explain why some species have common traits. The most likely explanation for the presence of a trait that was not present in the ancestor of the whole group is that it evolved from a more recent ancestor.
Two extensive groups of analyses exist to examine phylogenetic relationships: Phenetic methods and cladistic methods. Phenetic methods, or numerical taxonomy, use various measures of overall similarity for the ranking of species. They can use any number or type of characters, but the data has to be converted into a numerical value. The organisms are compared to each other for all of the characters and then the similarities are calculated. After this, the organisms are clustered based on the similarities. These clusters are called phenograms. They do not necessarily reflect evolutionary relatedness. The cladistic method is based on the idea that members of a group share a common evolutionary history and are more closely related to members of the same group than to any other organisms. The shared derived characteristics are called synapomorphies.
The introduction of two important tools has dramatically improved the study of phylogenetics. The first tool is the development of computer algorithms capable of constructing phylogenetic trees. The second tool is the use of molecular sequence data for phylogenetic studies.

Phylogenetics can use both molecular and morphological data in order to classify organisms. Molecular methods are based on studies of gene sequences. The assumption of this methodology is that the similarities between genomes of organisms will help to develop an understanding of the taxonomic relationship among these species. Morphological methods use the phenotype as the base of phylogeny. These two methods are related since the genome strongly contributes to the phenotype of the organisms. In general, organisms with more similar genes are more closely related. The advantage of molecular methods is that it makes possible the study of genes without a morphological expression.
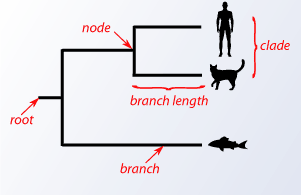
As previously mentioned, closely related species share a more recent common ancestor than distantly related species. The relationships between species can be represented by a phylogenetic tree. This is a graphical representation that has nodes and branches. The nodes represent taxonomic units. Branches reflect the relationships of these nodes in terms of descendants. The branch length usually indicates some form of evolutionary distance. The actual existing species called the operational taxonomic units (OTUs) are at the tip of the branches on the external nodes.
Tree construction methods
Some methods have been proposed for the construction of phylogenetic trees. They can be classified into two groups, the cladistic methods (maximum parsimony and maximum likelihood) and the phenetic method (distance matrix method).
Maximum parsimony trees imply that simple hypotheses are more preferable than complicated ones. This means that the construction of the tree using this method requires the smallest number of evolutionary changes in order to explain the phylogeny of the species under study. In the procedure, this method compares different parsimonious trees and chooses the tree that has the least number of evolutionary steps (substitutions of nucleotides in the context of DNA sequence).
Maximum likelihood This method evaluates the topologies of different trees and chooses the best based on a specified model. This model is based on the evolutionary process that can account for the conversion of one sequence into another. The parameter considered in the topology is the branch length.
Distance matrix is a phenetic approach preferred by many molecular biologists for DNA and protein work. This method estimates the mean number of changes (per site in sequence) in two taxa that have descended from a common ancestor. There is much information in the gene sequences that must be simplified in order to compare only two species at a time. The relevant measure is the number of differences in these two sequences, a measure that can be interpreted as the distance between the species in terms of relatedness.
Molecular phylogeny was first suggested in 1962 by Pauling and Zuckerkandl. They noted that the rates of amino acid substitution in animal hemoglobin were roughly constant over time. They described the molecules as documents of evolutionary history. The molecular method has many advantages. Genotypes can be read directly, organisms can be compared even if they are morphologically very different and this method does not depend on phenotype.
Phylogeny is currently used in many fields such as molecular biology, genetics, evolution, development, behaviour, epidemiology, ecology, systematics, conservation biology, and forensics. Biologists can infer hypotheses from the structure of phylogenetic trees and establish models of different events in evolutionary history. Phylogeny is an exceptional way to organize evolutionary information. Through these methods, scientists can analyse and elucidate different processes of life on Earth.
Today, biologists calculate that there are about 5 to 10 million species of organisms. Different lines of evidence, including gene sequencing, suggest that all organisms are genetically related and may descend from a common ancestor. This relationship can be represented by an evolutionary tree, like the Tree of Life. The Tree of Life is a project that is focused on understanding the origin of diversity among species using phylogeny.
References:
1) Whelan S., Lio P., Goldman N., (2001)Molecular phylogenetics: state-of-the-art methods for looking into the past Trends in Genetics, Volume 17, Issue 5, 1, Pages 262-272
2) Berger J. Introduction to Molecular Phylogeny Construction. BIOL 334.
3) Wen-Hsiung Li. Molecular Evolution. Sinauer Associates, 1997.
4) Pagel, M. (1999) Inferring historical patterns of biological evolution. Nature 401, 877–884
5) Zuckerlandl, E. and Pauling, L. (1962) Molecular disease, evolution, and genetic heterogeneity. In Horizons in Biochemistry (Kasha,M. and Pullman, B., eds), pp. 189–225, Academic Press 1921–1930
6) Felsenstein, J. (1981), Evolutionary trees from DNA sequences: a maximum likelihood approach, Journal of Molecular Evolution 17:368-376
7) Endo T., Ogishima S., Tanaka H. (2003) Standardized phylogenetic tree: a reference to discover functional evolution J Mol Evol; 57 Suppl 1:S174-81. Plant Species Biology
8) Murren C. (2002) Phenotypic integration in plants. Plant Species Biology. Volume 17 Issue 2-3 Page 89
9) Tree of life web project. What is phylogeny?
10) National Center of Biotechnology Information. Systematics and Molecular Phylogenetics.
11) Embley M. Molecular Systematics and evolution of microorganisms.