ONCOGENES: THE (AUTOSOMAL) DOMINANT EVIL
(August 2003)
In the beginning, there were chickens
Surprisingly enough, our understanding of tumour formation has its roots not in humans, but in the chicken. For the past century it has been known that viruses can be causative agents of cancer. Cancer-causing elements were first described in viruses infecting poultry in 1909 [1]. Later, it was shown that the injection of these viruses was sufficient for tumour formation. But the molecular basis of cancer was unearthed only with the discovery of cellular homologues to these cancer-causing elements in 1976 [1]. When the viral DNA sequences were isolated and characterized they were found to have high similarity to genes that already existed in the animal. These genes were named proto-oncogenes, and their viral, cancer-causing counterparts were called oncogenes.
Discovery of similar viral and cellular DNA sequences inspired a comparison of the two, revealing particular mutational differences. The hypothesis that the virus had at one point incorporated host genomic DNA into its own genome was suggested. This hypothesis postulated that these DNA sequences encoded for proteins involved in cell proliferation. By incorporating these genes and mutating them, the virus increased host cell proliferation, and consequently, its own replicative potential. This hypothesis led to the prediction that mutated forms of these same genes would likely be found in naturally occurring tumour cells as well. Scientists examined tumour cells for such genes, and transfected tumour DNA into normal fibroblasts to determine if the introduction of these mutated genes was sufficient to cause cancer. Indeed, they were, and awareness of the cellular oncogene was born [1].
Change a proto-oncogene to an oncogene and voila!
Oncogenes are mutated genes that contribute to cancer development by disrupting a cell’s ability to control its own growth and DNA repair mechanisms2,5. Normally mitosis (cell division) is a carefully regulated event, requiring the activation of one protein to activate another, in what is known as a signal transduction cascade. This cascade eventually culminates in changes in gene expression that prepares the cell for mitotic events.
Oncogenes are mutated forms of proto-oncogenes; these genes are often involved in growth signalling and anti-apoptotic pathways [2,5]. When proto-oncogenes mutate to become oncogenes they retain their functionality, but are no longer capable of responding to normal regulatory signals (see Figure 1).
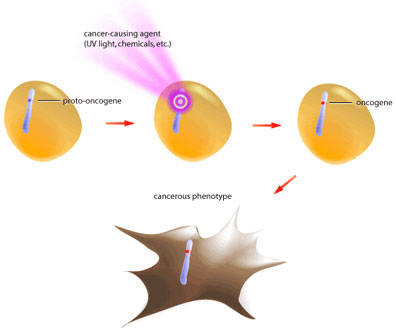
Such is the case of the growth factor receptor egfr (epidermal growth factor receptor). In its proto-oncogenic form it requires binding of the growth factor molecule to enable its kinase activity [3]. The catalytic domain then proceeds to transfer phosphate groups to its target proteins to activate them, which ultimately leads to translation of proteins involved in mitosis. The oncogenic form of egfr produces a receptor that does not require binding of growth factor, but instead is constitutively active (see Figure 2). In this way the oncogene product is capable of always activating the pro-growth pathway in the absence of pro-growth signals. Also, oncogenic mutations are dominant because only one mutated allele is necessary to confer the cancerous behavior [5]. At present, over 100 oncogenes have been identified in various cancers [2].
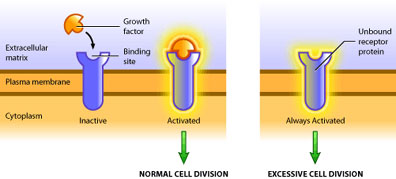
There are several ways in which oncogenes can promote the formation of a tumour. A cell’s growth is limited in two ways: 1) inactivation of the growth pathway and 2) activation of the apoptotic or differentiation pathways [6]. A tumour is only able to develop once it has found a means to bypass these two pathways. Hence, oncogenes are typically mutated forms of regulatory proteins within these pathways. This includes over-production of a growth factor, constitutively active receptors or intermediary molecules, or increased expression of proteins that inhibit apoptosis. No one oncogene itself results in cancer – cancer requires several mutations [6]. But one oncogene can increase cell division, and consequently, increase the probability of other mutations occurring, which can eventually culminate in cancer.
How does a proto-oncogene become an oncogene?
An oncogene can be the result of several different types of DNA alterations. These fall into two broad categores: point mutations and chromosomal rearrangements. Point mutation refers to the substitution of one base for another, resulting in a change in that particular amino acid. In contrast, chromosomal rearrangements involve whole pieces of chromosomes breaking off and fusing with other chromosomal fragments. Both of these events can result in either altered activity or intracellular levels of the protein (see Figure 3).

Point mutations most commonly produce a protein that has lost its ability to be regulated by external signalling, as is the case of egfr. The point mutation translates into a changed amino acid that has different biochemical properties than the amino acid the DNA sequence originally encoded for. This change in amino acid can mimic phosphorylation, or inhibit binding of negative regulatory molecules, resulting in a constitutively active protein [5].
Chromosomal translocations can produce oncogenes in several ways. Firstly, the fusion of one chromosome to another can result in a strong promoter being placed upstream of a gene that would normally be either absent or present only in very low quantities. This has the effect of increasing the intracellular concentrations and activating pathways that would normally be silenced.
Secondly, chromosomal translocations can result in fusion proteins, or proteins that are the product of two separate genes that have been joined. Chromosomes often endure a break in their coding regions — when two chromosomal fragments fuse, the genes that the breaks occurred in become one coding sequence that is transcribed and translated as one protein. Fusion proteins can be oncogenes for a number of reasons. If the fusion protein is the product of a highly transcribed protein, then expression of the fusion protein will be high. If the second portion of the fusion protein is a molecule involved in signalling that would normally be present in very low concentrations, then its sudden increase in expression as a result of being a fusion product could increase its activity.
Conversely, if a chromosomal break in a gene for a signalling protein results in the removal of its regulatory domain, then the fusion protein will consist of a molecule with signalling capacity that has no way of being negatively regulated. Such is the case of the Bcr-Abl fusion protein. Abl is a tyrosine kinase that requires cytokine stimulation to be activated. In its monomeric form Abl is inactive [4,5]. When cytokines are present, two monomeric Abl proteins bind one cytokine molecule – this close proximity allows the proteins to dimerize and auto-phosphorylate, resulting in activation. Conversely, the bcr gene contains a dimerization motif, but no kinase activity. When a translocation occurs between chromosome 9 (containing the abl gene) and chromosome 22 (containing the bcr gene), the fusion product contains the dimerization domain of Bcr and the kinase domain of Abl. Consequently, the fusion protein dimerizes in the absence of cytokine, resulting in a constitutively active tyrosine kinase and uncontrolled cell division.
The relevance of oncogenes…
After years of research and numerous attempts to cure cancer, there is still a gaping hole in our current understanding of the disease. Many oncogenes have been discovered within the last two decades. The role of oncogenes in oncogenesis, and how the proto-oncogene is mutated to give rise to this aberrant protein has been unearthed. But what clinical implications does our newly found knowledge have?
A greater understanding of oncogenes represents new ways to treat cancer. Identifying new oncogenes allows for a new means of diagnosing cancer. Further characterizing the activity of oncogenes provides us with new drug targets for effectively treating cancer. Eventually this reservoir of information may pave the way for gene therapy, and thus eliminate the occurrence of cancer. Each new piece of information gathered potentially provides us with another strength against this disease.
References
1. Weinberg, R.A. One Renegade Cell: How Cancer Begins. New York: Basic Science, 1998.
3. Carpenter, G. et al. (1991). Activation of second-messenger pathways by epidermal growth factor. In Brugge, J., Curran, T., Harlow, E. and McCormick, F. (Eds), Origins of Human Cancer (pp 255-263). New York: Cold Spring Harbor Laboratory Press.
4. Witte, O.N. et al. (1991). Role of the BCR-ABL oncogene in the pathogenesis of Philadelphia chromosome positive leukemias. In Brugge, J., Curran, T., Harlow, E. and McCormick, F. (Eds), Origins of Human Cancer (pp 521-524). New York: Cold Spring Harbor Laboratory Press.
5. Tannock, I.F. and Hill, R.P. The Basic Science of Oncology, 3rd ed. New York: McGraw Hill Companies Inc., 1998.
6. Hanahan D, Weinberg R.A. (2000). The hallmarks of cancer. Cell. 100: 57-70.
(Art by Jiang Long – note that high res versions of image files available here)